
Transcript featuring Gary Patti (Panome Bio) and Dipender Gill (Sequoia Genetics)
DIPENDER
So Gary, great to get a chance to talk again. I know we’ve had numerous really stimulating conversations about proteomics, genetics, the intersection of science, and how these advancing technologies can be leveraged towards generating translational insights to benefit patients. I think we’re going to spend the next 30 minutes or so discussing recent developments, how we see those, and our opinions and reflections on how the field is moving. Maybe we might kick off by introducing ourselves. I’ll let you go first, Gary.
GARY
Yeah, it’s a pleasure. Thanks, Dipender. I’m Gary Patti. I wear a couple of different hats. I’m a professor at Washington University in Saint Louis where I’m in the departments of Chemistry, Genetics, and Medicine, and I’m also the CSO of Panome Bio, which is a multi-omics company that does various kinds of profiling as a CRO service.
DIPENDER
And by background I’m a physician specializing in pharmacology and internal medicine, and I’m also an academic whose research focuses on leveraging human genetic data for drug discovery and development. And I’m the CEO of Sequoia Genetics, which is an R&D consultancy that leverages human genetic data for improving the efficiency of drug development. So to kick off, Gary, let me start with a very basic question — and I think it’s an important question for starting at the foundations. What is proteomics, and what does that mean in the modern R&D setting?
GARY
Yeah, it seems initially like a straightforward question, but some of these terms become buzzwords and then get appropriated by various technologies and can become a little ambiguous. Historically, when you attach the word “ome” to something, it means you’re measuring all of something. So if you say the genome, it indicates that you’re profiling all of the genes. And by analogy, if you say the proteome, that means you’re assessing all of the proteins. And what makes it a little ambiguous — even though it seems like a softball question — is that not all the technologies measure all of the proteins, so technologies deemed to be proteomics technologies aren’t necessarily measuring all of the proteins. But put simplistically, the idea is really to measure a lot, or most, of a particular type of molecule. I think most people are probably familiar with genomics — the idea being that there might be a gene that is altered in a particular class of people that provides some signature, perhaps of a diagnosis, or prognosis, or to inform treatment. And proteomics by extension is really the same sort of idea, but instead of looking at genes that serve as signatures, you’re looking for proteins. And just to clarify, maybe adding one additional point for those folks who are familiar with genomics but perhaps less familiar with other types of molecular signatures: you can really think of a cell as containing three types of molecules. You have genetic material, which would be covered by genomics. You have small molecules, which would be your metabolites, lipids, drugs, and so forth. And then you have proteins. Proteins are macromolecules — they’re typically large, so we’re looking at kilodalton size — and it’s important to make that distinction because they are different from lipids and metabolites.
DIPENDER
That’s very clear. So the very basic understanding is that the proteomic technologies that are widely publicized and spoken of — those wouldn’t pick up metabolites and those wouldn’t pick up genetic molecules. They would specifically focus on proteins, right?
GARY
Yeah, exactly. And of course you can do multiple omics — that’s the area of multi-omics, which has become incredibly popular and provides additional insight by layering more technologies. You don’t have to just do proteomics. In fact, there’s a lot of evidence that doing proteomics with genomics, or proteomics with genomics and metabolomics, provides even more insight. But you’re right, they are different technologies with different methods to conduct the actual experiments.
DIPENDER
OK, that’s very clear. From my own experience, I understand why genetics and genomics is important — genes code for proteins, proteins make up the majority of drug targets, particularly biologics and small molecules, and therefore by studying naturally allocated genetic variation we can infer the causal effects of these protein drug targets. And so we can prioritize therapeutic opportunities, inform heterogeneity and potential biomarker effects, and really leverage nature’s randomized control trial of randomly allocated genetic variation to inform drug discovery and development efforts. That paradigm has been well described for more than two decades, and certainly over the last five years we’ve seen an explosion in the availability of genetic data and therefore the application of Mendelian randomization paradigms to infer causal effects of drug targets. So I follow in my career why genetics is important. How has proteomics evolved in the last five to ten years, and why do you think it’s important?
GARY
So there are really two important pieces there. One is how it’s evolved in the last five years, which I think is inseparable from the second part of your question — why is it important? Because of course something can be important, but if it’s not accessible or feasible, it’s not useful. So let me put a pin in the first part and say that there’s been a lot of evolution over the last ten years that has made proteomics now accessible at large scale. Because to do the kinds of analyses that you’re talking about — by analogy with genomics — you need a lot of data, and you have to be able to acquire a lot of proteomics data from a lot of people. That’s an important criterion, but I think we should come back to that later because there’s actually quite a lot to tease apart. The second part — why is it important — is also very key, because why would we attempt to do something that’s not important? The paradigm really comes down to the central dogma of biology: genes encode proteins, and proteins then act as the enzymes that transform metabolites and lipids. While genes undoubtedly provide incredible insight, there are complications — you can have a dysregulated gene, or alterations in genes that don’t necessarily translate to a phenotype; perhaps a gene is not expressed, or whatever it may be. So I think one compelling reason to move downstream from genes is that you can potentially look at things that are a little closer to the phenotype. If you find, for example, changes in protein levels, that tells you more about what’s happening than genetics information alone — especially if you’re overexpressing a protein, or modifying it with what’s called a post-translational modification, which we’ll talk about later. And you might ask, well, why stop there? I mentioned the central dogma — you’ve got genes at the top of the cascade, proteins catalyzing reactions, and then the substrates and products which are metabolites. That’s one of the reasons why metabolomics and lipidomics are also worth looking at with omics technologies. But I think what makes proteomics really ripe, as a metaphor with temperature, is this: genomics you can think of as maybe being a little too cold. Metabolomics, you can think of as perhaps a little too hot, because it’s very sensitive to environmental changes — what you eat, when you last ate, how much sleep you get, your environment — it changes very rapidly and is very transient by its nature. The proteome is in the middle. If the genome is too cold and the metabolome is too hot, you could perhaps think of the proteome as being just the right temperature to interrogate. So I think you could build a compelling case for why it’s important to be thinking about proteins in addition to genes.
DIPENDER
So let’s take a step back and I’m going to play devil’s advocate, because I think it’ll be fun. With genetics there are two clear advantages for improving the paradigm of drug development: first, it’s human data, so we infer causality in humans; and second, exactly that — the random allocation of genetic variants. With that paradigm we’re overcoming two of the major limitations in drug development: we’re not relying on animal models, and we’re not relying on observational associations — we’re inferring causality. So we have causal effects in humans. Now with proteomics, metabolomics, and lipidomics, are we still not falling down to one of those hurdles? These are associations — they don’t infer causality, and therefore we can’t disentangle confounding or reverse causation.
GARY
Yeah, I mean, inferring causality is always going to be tricky in human studies at the epidemiological scale — undoubtedly that’s a challenge. I do think looking at something through a multi-omics lens provides a lot more interesting data and in some cases more clarity on what’s going on. One of the ways I like to think about it is with an American football analogy. When they record an NFL game, they use something like twelve-plus cameras filming from different angles — an aerial view, one from the middle of the field, all these different perspectives. Whenever a play happens and they can’t quite determine what occurred, they appeal to all of those different views. There’s the main view you see on live television, but then you have all these backup views, and when it becomes too close to call, they appeal to them. I think in many ways that’s what we’re doing with multi-omics. You’re looking at biology through one lens, one camera angle, and sometimes it’s difficult to tell what’s going on. But if you have these other camera angles to appeal to and you say, “my hypothesis from this view is that this particular thing is happening,” and then you look at it from these other angles and gain additional evidence — that helps support the causality framework. But I would agree that no matter what you do, if you want to derive mechanistic information, it’s in my opinion very difficult to get away from animal studies in some settings, or from natural experiments where you look at an intervention and see if something changes. I think in general those controlled experiments are very hard to do, and at the epidemiological population scale, it is challenging to get at mechanism regardless of what science you’re doing at the human level.
DIPENDER
Let’s add a couple more spices to the cooking pot then. Let’s talk about doing omics — proteomics, lipidomics, metabolomics — in a clinical trial setting, and let’s talk about this emerging phenomenon of proteogenomics. Starting with the former: if you have omics in trials, you can infer causality because you have a randomized intervention and you’re exploring the causal effect of that intervention on the proteomic or lipidomic profile. So you can infer causal effects and you have biomarkers of target engagement or mechanism of action. You can say these omics are changing because of our intervention compared to placebo or another intervention. And similarly with proteogenomics, where you have random allocation of a genetic variant that mimics an intervention — you can say this is causality because we have randomization. For me, those are some of the most exciting applications of proteomics, and they’re becoming more and more popular for overcoming the lack of translatability of animal studies while also inferring causal effects. What’s your experience of these, and what do you think is really limiting the opportunity with implementing these technologies?
GARY
That’s a great question. What I was saying earlier was really just about looking at existing data when it’s hard to extract mechanism. But I see what you’re saying — if you’re intervening, doing a clinical trial, setting up something and actually changing diet, a drug, or what not — I concur with what you’re saying. A good example that’s getting a lot of attention right now is tau measurements in Alzheimer’s — the fact that this provides a clue of something that is a marker in the blood that changes perhaps well in advance of the actual disease phenotype. It’s a really beautiful example because if you’re not showing any cognitive impairment yet, it’s very hard to say whether you should be doing something. Alzheimer’s is a condition that takes a while to develop — we’re talking about a decade or two decades over which time this pathology starts to do its damage and lead to the typical consequences. So you’re seeing questions like: if a person feels perfectly healthy but their tau is elevated, can we intervene and make that tau go down? And will that provide possibilities for being able to treat people for a disease whose phenotypes aren’t necessarily established yet? I think that sort of approach is extremely appealing. But the challenge, as you point out, is making the measurements first — finding what those markers are. I’ve just named one example, but there could be hundreds or thousands. So how do we find them? And once you find them, how do you measure them in a way that’s reproducible, with enough sensitivity, from the right sample matrix, with all the questions that come up around profiling coverage? So there are really two sides to that coin: how do we find what to look at, and how do we measure all of those things — both to find the markers and to monitor them after we know what’s important?
DIPENDER
Yeah, so I think that’s a really good point. And it brings us back to one of the first statements you made, which is around the definition relating to omics — essentially covering everything. So to bring it back to that point: do you think these technologies should be targeted, or do you think they should be untargeted? Because you don’t know what you don’t know. There may be things out there that are critical for the causal mechanism that we otherwise wouldn’t capture because we’re not yet fully aware of them. But there’s a trade-off, right? The more things you measure, the more complexity, the more cost, the more multiple testing correction there is, and there are power and sensitivity issues. Where is the sweet spot, and how is that evolving over time as the technology advances?
GARY
Yeah, I think that’s the key question, and I would argue it’s context dependent. There will be cases where I think you should absolutely be measuring as many things as you can at a global scale. If you’re at a hypothesis-generating phase, that’s the best way to do it — I always say you want to play as many lottery tickets as you can. Why would you buy five lottery tickets when you can play ten? The more lottery tickets you have the better — but lottery tickets cost money, so if you can only afford five, you can only afford five.
DIPENDER
No, it’s a good point. But I want to contrast that paradigm — that stage of a research project — to the targeted analysis like the tau example, where you already know the target and you’re looking at interventions and testing that one target. You don’t need to do untargeted there. Now you could say, well, I’m going to layer on untargeted because I want to see if there are other things that change as well, and perhaps there are subsets of cohorts you want to try to dissect out. So maybe having those, but —
GARY
And so maybe having that broader layer, but typically the way a project evolves in my mind is that you start with a really wide net and try to figure out what you’re looking at, try to infer some correlation. And then as you explore causation and ways to interfere or intervene, then you look in a more targeted sense at what those markers are. And I think what you said is so important, because there is a relationship between the number of things you measure and how well you can measure them. We have this conversation all the time, and I have this story from early in my career — a very important person approached me and wanted to do omic-scale measurements, but really only cared about one particular molecule. Instead of saying “I really only care about this one molecule,” they came and said “I want to do this untargeted.” Their thinking was, “you’re going to give me everything anyway, so I want all that stuff and then I’ll get the molecule I care about — all that other stuff is essentially free.” And ultimately after many years of working together, they were able to pull out the trend they expected for the molecule they were interested in. And they said, “that’s it, that’s all I wanted to see.” And I don’t think that’s the right approach if you really only care about one thing. It’s usually better to just focus on that or a couple of things, because as you point out, there are disadvantages to measuring a lot of things. Broadly across all technologies, I would say the more things you measure, the less well you measure them. If you really care about one thing and you want to measure it as well as you possibly can, it’s better to just measure that one thing and not try to measure fifty thousand others, because those fifty thousand will come at the expense of lower data quality for the thing you actually care about. So I think you really have to be honest with yourself and ask: where in the project are we? What are we looking for? Are we hypothesis generating, or do we have a strong hypothesis we want to test and validate that involves a limited number of targets?
DIPENDER
Really, really powerful points. I want to bring us towards another theme — we’ve started alluding to it — things like cost, expense, and the “M word.” I kind of get the sense that money is a dirty word in academia. We’re purists; we’re just interested in science. But I think the reality is it’s a trade-off between the resources we’re putting in and the societal value we’re generating. A good strategy is to maximize societal value for the same denominator of resources allocated. What I’ve observed in the field of genetics is that the explosion and excitement we’re experiencing recently is because of two factors largely related to money: one is the reduced cost of genotyping, which has gone down exponentially over years, and secondly the reduced cost of data storage. Taken together, that means we can generate, store, and analyze more data on more people related to more biomarkers and phenotypes, and we can really scale exponentially the complexity and value of the analysis we’re doing. How has the evolution of omics mirrored this, and what implications does that have for modern day analysis?
GARY
Yeah, you’re referring to proteomics specifically?
DIPENDER
Yeah. Well we can start with proteomics, but the field generally.
GARY
Yeah, let’s start with proteomics. I think it’s a really interesting case because proteomics has been around for a long time. The origin of proteomics isn’t that distant from the origin of genomics — it wasn’t long after people started thinking about sequencing the human genome that people started thinking about measuring the entire proteome. So it’s been around for a long time, but for many people the advent of proteomics is much more recent. The reason for that is the technologies over the last handful of years that have emerged — new, innovative technologies that were not available previously. Initially, proteomics was really conceived as a mass spectrometry field. What I mean by that is we were using instruments called mass spectrometers to measure as many proteins as we could at the global level. And while that is exciting and intriguing and an integral piece of the field, in many ways it was complicated for people for several reasons. The technologies weren’t quite ready to do the kinds of population-scale studies that we’ve already discussed here today. Breaking it down briefly, just for context: what does a mass spectrometer look like, how much does it cost, and how does it run? Mass spectrometers are instruments that are essentially scales. The analogy I always use is a bathroom scale — you get on it and it gives you a measurement of how much you weigh. If you and I both got on a scale, we would have different measurements. You could imagine creating an encyclopedia where every time somebody stood on the scale you could record it in a database, and then if you ever saw that number on the scale again, even without knowing which person you were looking at, you’d say “oh, that’s the number I know is associated with this person.” That’s essentially how a mass spectrometer works. It’s a scale, but it measures things at a microscale that we can’t see — you can’t see a protein with the naked eye, but a mass spectrometer can measure its mass. The mass of any given molecule doesn’t change, so in some ways it’s like a fingerprint — you can always identify that same molecule because its molecular mass fingerprint doesn’t change. The mass of a stable compound should not change. And so that’s what a mass spectrometer essentially does. It measures the mass of anything you put in it — it could be a protein, but it doesn’t have to be. There’s a whole field of petroleum analytics that measures types of molecules associated with petroleum. So you can use it for all sorts of things. It’s a very agnostic, broadly applicable type of device, and the early days of proteomics were really conceived using mass spectrometry technologies. The downside is that mass spectrometers are complicated pieces of equipment. They come in variable sizes, but they’re usually about ten feet in length and five feet wide — about the size of three or four centrifuges on a bench. Some instruments can be larger, floor-standing instruments that come all the way to the ceiling, and some weigh five hundred pounds. They range in cost from a couple hundred thousand dollars for lower-end instrumentation all the way to two or three million dollars for the highest-end commercial instruments. It’s like cars — there’s a range, and some are really nice and do a bunch of fancy things while some just get you from A to B. Because of the cost, the size, and the complexity required to run them, there’s been more of a trend recently to design mass spectrometers that are more accessible and can be run without extensive training. But historically, graduate-level or at least extensive training was required to operate a mass spectrometer. Because of that, and because of limitations on throughput, that cornered proteomics into a relatively small part of the universe, and clinicians like yourself probably weren’t very familiar with it or weren’t thinking about it seriously as a disruptive technology that could impact healthcare. What changed a handful of years ago was the advent of a couple of other different technologies that we now broadly refer to as affinity-based proteomics technologies.
DIPENDER
And so affinity-based — because my commercial experience of these is twofold, right? There’s the aptamers and the antibodies. Are those both affinity-based proteomic measures?
GARY
Yes, I’d want both of those together.
DIPENDER
And just for my understanding, what’s an aptamer, what’s an antibody, and how do they differ even though they’re both affinity-based?
GARY
Yes, they’re really both the same approach — the difference is how you interact with a protein. There are two different companies that have pioneered two different technologies. The way to think about what affinity technology is trying to do by its very definition: imagine a sample like a cell, which contains thousands of proteins. What affinity technology is trying to do is pull down a specific protein — separate, isolate, or purify it in some way so that you can assess how much of it is present. We’ve done that for a long time. Most people are familiar with using antibodies in the lab; there are all sorts of antibody tests. The key difference is whether you’re using amino acids as the basic unit to build something that will pull down a protein, versus using nucleotides as the basic unit. You can debate which is better — which is more specific, more sensitive, and so on — and I think the verdict is still out. You can make cases that they’re complementary: maybe one protein works better with one assay and a different protein with another. But the bottom line is really that you’re trying to differentiate one protein from a sea of other proteins, with the goal being that once you purify or isolate it from its other companions in the cell or biological sample, you can then assess how much is present. These technologies can also take advantage of sequencing to assess how much protein is present — by doing quantitative measurements counting how often the binding occurs on a genetic sequencer of the same type used for genomics experiments. What this did was take something that was a black box — proteomics on a mass spectrometer, which people thought was complicated or hadn’t heard of or felt was too sophisticated — and bring it to a technology that most biologists, clinicians, and scientists had already heard of: antibodies, aptamers, sequencing. These are much more familiar to the majority of scientists than mass spectrometers. And so because of that, it led to a boom, because they developed libraries of affinity reagents. You could imagine developing an affinity reagent for protein X, another for protein Y, another for protein Z, and pulling down each one of those proteins individually. Normally we think of antibody tests as very low throughput, doing one at a time — but essentially this is like doing antibody assays on steroids, pulling down all of these different proteins at a comprehensive scale and then assessing how much is present. These two opposing but complementary technologies emerged a handful of years ago and immediately drew a lot of attention, because for many scientists this was the first time proteomics was accessible. So although you could go back and find papers from the 1990s where people were talking about proteomics and its impact on healthcare, this affinity-based proteomics was in my opinion really a catalyzing agent that brought that technology to the sphere of medicine in a very important and unique way that changed the landscape of proteomics.
DIPENDER
So from my understanding, what was the transformative factor? Was it that they started using technologies people were familiar with so people understood it better? Was it that these technologies brought the cost down? Or was it the popularization of the methodology and the communication of its potential importance? What was the secret sauce that led to this explosion in the field of proteomics and the commercial extent that we see today?
GARY
Yeah, I think all of the above, honestly. It’s hard for me to pinpoint the coefficients of all those reasons, but I think every one of them goes kind of hand in hand. If there’s a lot of marketing around a technology you’ve never heard of, that’s a much heavier lift. But if you’re starting to see and hear about a technology everywhere, and you understand it because all of its composite pieces are things you’re familiar with, it tends to land and stick a little better. So I think it really is a little bit of everything, but perhaps one factor that I think is among the most defining is that the types of things those assays could do, mass spectrometers were not well positioned to do at the time. It was filling a void that was really currently inaccessible with any other technology — and that is: can you measure hundreds, thousands, tens of thousands, or hundreds of thousands of samples? While mass spectrometry proteomics has come a long way, at the time trying to do those sorts of comparable experiments on mass spectrometers was a much heavier lift than the way they could be done on the affinity platforms. So it was really a unique constellation of factors including all the things you mentioned, but I would add one more: it had the capability to scale in ways that mass spectrometers at the time really couldn’t. Now, I’ve been drawing this historical picture out quite a long time, but hopefully it’s providing some color to how you think about the field. What this open market dynamic did, in my perspective, is really draw the vendors of mass spectrometers to try to address this void. It became quite apparent that there was high demand — people were adopting these affinity-based technologies readily, there was a surge and a lot of uptake. And the mass spectrometry companies, in many ways, looked at that and said, “if we could make some improvements on this technology for scale and throughput, we could bring ourselves into that same spot where those kinds of experiments would also be achievable by mass spectrometry.” And that’s exactly what happened. There were a couple of mass spectrometers developed over the last handful of years — from companies like Bruker and Thermo — that were geared at doing large-scale, high-throughput proteomics with mass spectrometers, making those aspects competitive with the affinity technologies.
DIPENDER
So let me understand a little bit better, Gary, because you’ve explained the affinity-based technology very clearly — that it was scalable, accessible, and all of those things led to that trajectory. How was mass spectrometry able to catch up? What changed? You said it was around for ages, it wasn’t able to scale, it didn’t popularize — what changed?
GARY
Yes, so I think what’s really changed — and there are a couple of different technologies, and each of the vendors has approached this differently. There’s a handful of mass spectrometry vendors, much like cars — there’s not an unlimited number of manufacturers of these instruments, and each has tried to come up with technologies to address these issues. The problem they were really trying to address is throughput. When I was still a trainee, to do a proteomics experiment with mass spectrometry, there was a popularized workflow called MudPIT, and doing that kind of experiment would take ten to twenty hours per sample, because it was really slow.
DIPENDER
Was that because you were a postdoc and were slow, or just that the technology was slow?
GARY
Touché. Well, I’ll answer that the same way I did before — it’s like a combination of factors. No, it was primarily the technology: the experiments themselves were just slow. If an experiment is going to take twenty hours, trying to imagine doing a thousand samples — or even a hundred samples — is really inconceivable. So you had to go from running samples over that time span to being able to run samples in, say, a few minutes each or an hour or so each. The other problem that comes up, which is very much a nuisance for mass spectrometry, is the dynamic range of the proteome — the spread in protein concentrations. One of the sample types people think a lot about is blood. When you look at human blood, a major portion of the protein content comes from a couple of very uninteresting proteins like albumin. One of the problems with mass spectrometry is that detection can be saturated — if you try to measure all of the proteins in human blood simultaneously, those very few but very abundant proteins end up taking all of the bandwidth of the detector and you can’t measure all of the low-abundance stuff that’s much more interesting. So in addition to throughput, which has been largely addressed by the manufacturers, there was also the development of front-end technologies that allow you to do pre-treatment before you run the mass spectrometry. That increases your dynamic range so that you can fractionate out really abundant proteins — either removing them or analyzing them separately so they’re not competing with really low-abundant proteins for detection. Both of those types of improvements have come a long way, which makes mass spectrometry much more suitable for clinical translational experiments and profiling assays.
DIPENDER
So Gary, with that in mind, where are we right now? How do the affinity-based technologies compare to mass spectrometry? Let’s talk about coverage, specificity, sensitivity, and the “M word” — how do they compare in terms of cost?
GARY
Yeah, let’s start with cost because I think that’s kind of a deal-breaker for many — I could imagine people turning the webinar off at that point. But I would say we’ve entered an era where the costs are really competitive. They’re all in really the same sort of ballpark, depending on how many samples you have and various other factors.
DIPENDER
Gary, is that a commercial factor? Is it that it costs the companies less but they’re pushing up the prices to maximize profit as long as it’s less than the competitor, or does it genuinely cost the same amount to run?
GARY
You know, it’s difficult for me to know — I’m not part of the organizations that sell the kits, so I don’t know what the manufacturing costs are. But I think this is a really interesting case of what the free market does to science. Proteomics mass spectrometry was around for decades, and the amount of change that’s happened in mass spectrometry proteomics since the affinity assays came online has been dramatic. You could say it’s happenstance, but it’s hard not to believe that competition in the market wasn’t a catalytic factor — when you have competition, it does amazing things. I think these companies are genuinely working hard to get the costs down, to develop cheaper kits, to find efficiencies. So I think you’ve seen a lot of different flavors of the ice cream.
DIPENDER
OK, so the technology has advanced and now because of the technological advances and competition in the market, the costs are comparable. What about sensitivity, specificity, and coverage — how do those compare across affinity measures? I don’t think we need to get into the details of different affinity measures; those companies publicize that enough themselves. But how does mass spectrometry compare to the affinity approach as an entity?
GARY
Yeah. And let me just clarify — when I say the costs are the same, I know someone might be watching this and say “yeah but I sent a quote to this company and it was $672, and this other company was $704.” I’m not saying it’s to the dime. I’m saying it’s in the same order of magnitude.
DIPENDER
Yeah, I don’t get hung up on that. So we’re not talking that one technology is two orders of magnitude different — you’re not adding an extra zero.
GARY
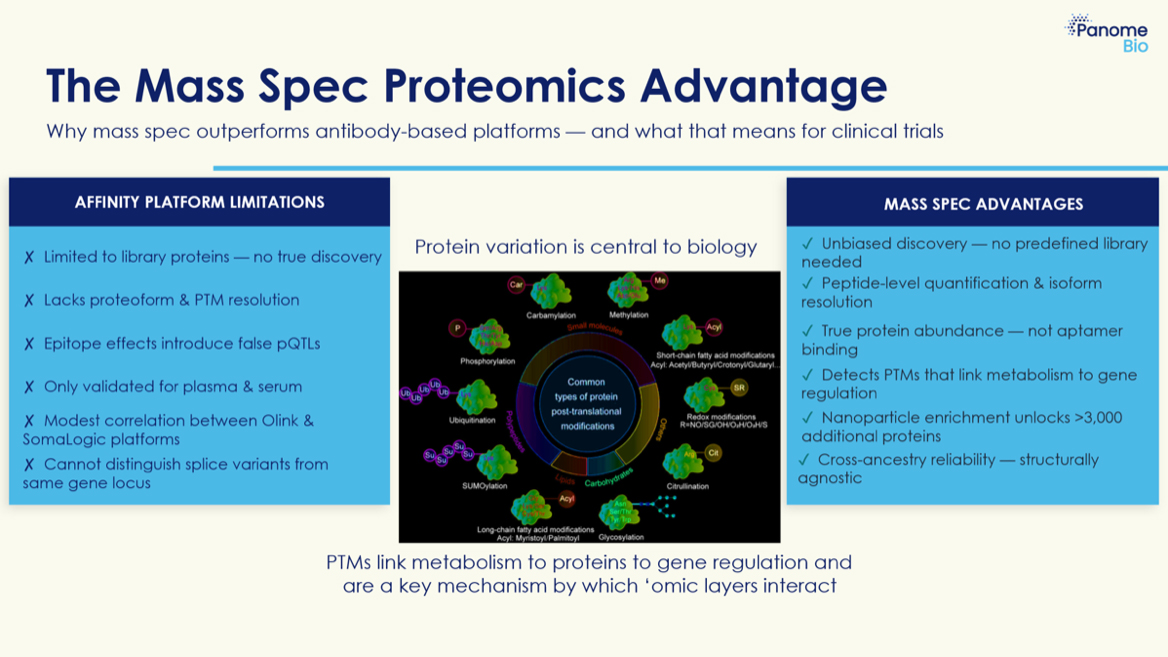
Yeah, let’s say the confidence intervals overlap. But to your question, the technologies are actually fundamentally, intrinsically different — and I think this is an incredibly important point when I talk with people about them. This is a point that is a little abstract and perhaps even a little philosophical. Dipender and I share this sort of philosophical interest and have thought about this more than most in this clinical/basic science space. So let’s take a swing at breaking it down. With affinity-based profiling, you can’t measure a protein unless you have an antibody or an aptamer that’s been developed to interact with that protein. So in the best case scenario, you develop an antibody or aptamer specific to protein X, and you pull down protein X, isolate it, and measure how much is there. But if you don’t have that antibody for protein X — let’s say there’s another protein, protein Y, and there’s no antibody, no aptamer, no affinity reagent to pull down protein Y — it is completely invisible to the experiment. So it’s really important to emphasize that affinity-based experiments can only measure things for which there are antibodies or aptamers designed to interact with those proteins.
DIPENDER
So hold on a second. What does that mean for isomers and post-translational modifications and glycation? HbA1c — glycated hemoglobin — is one of the most commonly clinically measured markers, right? What are the implications of those post-translational modifications?
GARY
I think that is such an incredibly important point. Could we put a pin in that? Because I think it adds another layer of complexity and I’d love to dive into that deeper, but the other question you asked was also so important and I don’t want to conflate the two responses — I think in many cases people see them as a little separate.
DIPENDER
OK, let’s take them in turn then, as you prefer.
GARY
OK. So for these affinity technologies, you have to have either the antibody or the aptamer or whatever affinity reagent you’re using to pull down the protein.
DIPENDER
Yeah, in simple terms: you can’t find what you’re not looking for. If you don’t have a sensor to find that protein, if you don’t have the affinity-based reagent, you’re not going to find it, right?
GARY
Yeah, exactly. The analogy I love to use — and I’ve sometimes included this cartoon in my presentations — there’s a gentleman on the ground crawling around looking for his wallet. A cop drives by and says, “what’s going on?” He says, “I’m looking for my wallet.” The cop starts looking too, and after a few minutes says, “did you lose your wallet here?” And the guy says, “no, I lost my wallet over in the park.” The cop says, “why are you looking here?” And he says, “this is the only spot there’s a street light.” So the same analogy applies here — if it turns out these are the proteins you want to look at, then great. But if the proteins you want aren’t included and there’s no antibody, they’re not going to show up. Now to the credit of the affinity-based companies, they have built large libraries. You’re looking at five thousand, ten thousand, fifteen thousand proteins — they have antibodies and aptamers for all of those. That’s a lot.
DIPENDER
How many proteins are there?
GARY
That’s a good question. And an even more relevant question is: how many proteins on their library will you actually measure in a sample? Because these numbers sometimes get conflated and they’re actually quite distinct. The number of antibodies or aptamers for different proteins that are present — that’s the maximum number of things you could ever conceivably measure, and that assumes that every antibody or aptamer designed for a particular protein actually finds that protein present in your sample. If you’re looking at something very simple with very few proteins, you won’t see all those signals. Or if you’re looking at some extremophile bacteria from a hundred miles under the ocean with all these unusual proteins, they’re not going to be on the panel. So you could imagine seeing very low numbers in that case. To your question about how many proteins there are — that seems like a softball question but it’s complicated, because it depends on how you define a protein. I’d like to return to this idea because one way to define a protein is using a term called a “proteoform.” A proteoform refers to different modifications on a protein, but let’s come back to that because it ties into the last question you asked about post-translational modifications. I’d like to first contrast what I just said about the affinity library to the way mass spectrometry works, because your earlier questions were so well-posed — they’re setting up the contrast perfectly. If we think about how a mass spectrometer works using the bathroom scale analogy: it doesn’t matter who stands on the scale. Someone you’ve never seen before could come in and stand on it — you still get a measurement. Anyone could come, you could place anything on your bathroom scale and you’ll get a measurement. And that’s the way mass spectrometry works. There’s no library required to measure the mass. You can measure, as I mentioned, petroleum-related compounds, food, lipids, metabolites — anything.
DIPENDER
The phrase I really like is: it’s untargeted, right? You’re not targeting anything specific. Whatever is there, you’re going to capture it.
GARY
One hundred percent. So it’s truly untargeted. And I think one of the complications is there’s been some ambiguity about “global” and “untargeted,” because in many ways if you’re doing truly untargeted and measuring everything, and global means measuring everything, then the words global and untargeted are synonyms. They’re both characteristics of a study. Only if we’re saying “omic” doesn’t that mean it’s global and holistic and untargeted? But anyway, that’s a side note.
DIPENDER
Yeah, no, I think — and this is so much fun philosophically. We could probably spend the next hour just talking about terms because there’s so much linguistics here: what the “ome” means, how terminology has evolved. But I think there’s actually a lot of this mismatching going on more broadly, not just with affinity technologies. If you have a panel that’s global, the perception is that it’s sort of untargeted because it’s so big. But if it’s not completely global — which unfortunately it’s not, you can never have a panel that covers absolutely everything — then even though it might be large, it’s not necessarily everything, which doesn’t equate with it being truly untargeted.
GARY
That’s very clear. So then let me ask you another question: is mass spectrometry global?
DIPENDER
That’s a good question too. Mass spectrometers are also not perfect instruments — there are going to be things that won’t show up. When you think about certain molecules, there are some that just don’t ionize well, meaning they just aren’t visible to a mass spectrometer. But in general, the amount of molecules that a mass spectrometer can measure is a very large, comprehensive panel. Saying it can measure absolutely everything in the universe would be inaccurate, but it can measure a lot.
GARY
Yeah. So then let’s go back to the question: how many proteins are there, and how does the theoretical coverage of the mass spectrometry paradigm compare to the affinity-based assays currently? And how do you see this evolving in the future?
DIPENDER
Yeah, so I think that gets into how you define a protein, and I’m going to put forward this term that’s often used in proteomics — and Neil Kelleher is one of the pioneers who really drove this forward. The term is “proteoform.” What is a proteoform? The way I like to think of it is a protein that’s been chemically modified. One analogy you can use: imagine you go to the store and buy a Christmas tree. Ten people might go to the same store and buy the same Christmas tree, but when they bring it home, they decorate it with different combinations of ornaments. You’ve got the ornaments your kids made, the ornaments from your relatives — everybody’s got a different combination. And even though they all started with the same Christmas tree bought at the same store, they all look different because they’ve been decorated in a personal fashion. And I’d say the way a Christmas tree is decorated has a big impact on what it’s achieving — the emotions it provokes.
GARY
Yeah, and by analogy that’s what a protein is — it’s chemically modified, and the way in which your protein is chemically modified can in many cases influence its function. We know, for example, in central carbon metabolism that most proteins are chemically modified, and those chemical modifications regulate some of the most important nodes of metabolic regulation. We call these post-translational modifications — “post-translational” because they occur after translation and you’re adding a chemical modification. There are actually quite a lot of different chemical modifications that can be added, and we don’t even know all of them yet. This is one of the really exciting areas of biochemistry right now.
DIPENDER
Sorry to interrupt — does that create a double-edged sword then? On the one hand I can see, hey, theoretically this could be a superior technology because it’s untargeted, you’re getting better coverage, you’re picking up these post-translational modifications. The concern I sometimes have with these large-scale approaches is that you get more data than you can make sense of — millions of parameters and you don’t know what to do with it. Sometimes simplicity is the ultimate sophistication. Just give me something I can handle, interpret, and make my life easy. How do we balance those two things?
GARY
Yeah, simplicity is great. I think the affinity technologies without question have been good for the field of proteomics — that’s absolutely undeniable. There are people for whom mass spectrometers were a black box, just too complicated, so far-fetched, with so many nuances that it wasn’t perceived as accessible. So I think you’re right, there has to be a balance. And the reality is there will be important discoveries made in both ways: there will be discoveries buried by complexity where a simplistic approach will uncover them, and there will be a lot of discoveries that require a complex approach because the underlying biology is in fact complex. So it really does get down to context. It depends on the problem, the question you’re trying to answer. My default always gets back to the lottery ticket thing — computationally, having complex data is something that can be manageable, and sacrificing that complexity because you’re worried you won’t be able to interpret it is maybe not the right sacrifice. But I’d love to hear your thoughts on that.
DIPENDER
Yes, I completely agree with that. I think if you have the data, you have the option. Say you have more data than you can handle — you simply put a filter on it and say, “this is too much for me to handle right now, but let me put a filter to look specifically at the things I want to look at.” And if I later want to do an exploration, I have the option to go back and look at the detail, the nuances, the minor changes, and the other subtle factors. But if I really want the basics, I just put a filter and it gives me the basics. You have that optionality. I much prefer that approach than being given something with no option to really understand the nuances or subtleties — the isomers, or how things in practice in biology might be exerting their effects. So I prefer that optionality. I think all information is good. It’s how you use it that differentiates its translatability.
GARY
And I 100% share that same perspective. The nice thing about the computational aspect is that it’s much easier to work with complex data computationally than at the analytical/technological instrument level. I can see how physical hardware adds a layer of complexity that can be very challenging. But when it’s computational and these are files that can be sent around, it’s much easier to work and partner with teams to address that complexity than, say, to build an instrument or do something in-house in your lab that requires physical hardware.
DIPENDER
Gary, I know we’re at the hour. We could talk for hours more. I’ve got a few other points I want to pick up very briefly — and if we need to shorten this later, we can. Where do you think is a better fit for omics: in trial data, in population-level cohort data, in selected samples — or are there nuances to each? A trial setting involves randomization of the intervention, so you’re asking what is the causal effect of the intervention. Cohort data has really exploded — UK Biobank, Our Future Health, All of Us, and the list goes on. How do you think the role of omics and proteomics should vary in these different settings, and how does that contrast with, for example, genetics?
GARY
I think in my perspective that’s exactly analogous to genetics, actually. The cases you put forward where genetics has been successful have involved both of those. I’d be curious to hear your perspective on whether you think that’s equally so across all of them, or if there’s one that stands out. But in my opinion, proteomics has an opportunity to excel across all of those possibilities, and they’re all complementary — one feeds the other. What you learn from large cohorts in finding important markers correlated with a particular disease state, and then being able to look at that marker with an intervention to see if you can change it, is a really good example of how those two could feed each other. But there are millions of different combinations and permutations of how you could design these studies. I would just say I don’t think there’s necessarily anything that makes proteomics especially unique in that regard — it really in many ways is analogous to genomics or other omics.
DIPENDER
Yeah, it’s an interesting point. I do think there are different roles for different settings. For example, in large-scale cohorts and consortium meta-analyses with hundreds of thousands of participants, I think genetics is far better powered to find genetic variation implicated in phenotypes and biomarkers. In contrast, proteomics in itself can be an epidemiological study — what is the association of the protein with different traits? You don’t necessarily need a hundred thousand participants for that, but sure, it’ll boost the power and allow you to look at subgroups and heterogeneity. Even with a hundred people you can say, “these traits are associated with these proteins.” So in my experience, genetics is great in the large-scale biobanks and consortia. Proteomics, if I had a choice between getting genetics or proteomics done in a trial — honestly I would pick proteomics. In a phase two trial of a hundred and fifty participants, proteomics is more likely to give you value than genetics. Genetics is great if you have a hypothesis-driven analysis, there’s some pharmacogenetic phenomenon, there’s a particular variant you need to stratify on. But undertaking a genome-wide scan even in a phase three trial with, say, two to five thousand participants — you’re very unlikely to make discovery efforts. It could be hypothesis-driven targeted analysis. So for me, genetics is more powerful in large-scale biobanks and consortium meta-analyses, while proteomics as an epidemiological endeavor can work better in trial settings. And then there’s the overlay of targeted pharmacogenetics where you say, “this genetic variant is affecting drug response,” and you can form subgroup analyses there. I do think there’s an interplay. The other related point is: genetics has been around for more than two decades, and I would argue that for the first ten to fifteen years it didn’t really deliver the promise people were expecting. That’s changed now because we’ve seen a massive explosion. Do you think — and this is the unknown, right — proteomics, I think we’d agree has exploded. Everybody’s talking about it, asking whether they should be getting it in their cohort or their trial. Do you think we’re at a point where people can definitively say this is the value of proteomics? If you look at genetics as a comparator, people can cite the statistics — targets with genetic support are two to three times more likely to succeed; if you have a genetic variant mimicking target perturbation, you can inform efficacy. The promise of genetics has proven itself where it’s been done the right way. Has proteomics proven itself yet?
GARY
Really intriguing question. The timescale, like you said — genomics has a ten- to fifteen-year head start. What we’re talking about with these technologies is much more recent, so certainly the number of successes is going to be limited just based on sheer time. But I think the first area that really comes to mind is what’s happening in the Alzheimer’s field, where you’re seeing some of these proteins that can be altered well ahead of an actual clinical phenotype. It’s a perfect example of what I was talking about with interventions, because then you can start to look at interventions. It’s very salient to Alzheimer’s because at least some of the thinking is that if you don’t try to adjust the pathology early, before the development of symptoms, it may be too late. And so if you can find a protein marker that signifies disease before there’s a clinical phenotype, and you can intervene and reverse that marker, rescuing that person from that phenotype — I think that would be just an incredible success story. We’re obviously not there yet, but when you look at what’s happening in the space, there’s a lot of momentum and a lot of promising results that are starting to appear that support some of that vision.
DIPENDER
Yeah, and I can say from my own personal experience looking at proteomics in cohort settings — UK Biobank being one example — and looking at proteomics in trial settings, some of which is published in the public domain, there is clearly value. You can look at proteomic profiles to examine heterogeneity of effect, predictors of effect, potential novel indication mapping, and adverse effects. So for me it’s proving itself. Now, the proof of the pudding is a phase three clinical trial — have the proteomics prospectively informed decision making? Has that proven itself? That’s going to take five years to come to fruition. Last thing to finish, Gary: if you were the director of a large biobank or a consortium effort or cohort study, or if you were a pharma executive designing how best to optimize the value and learnings from your clinical trials — going back to our fundamental strategy of maximizing the allocation of resources for generating the most societal benefit — how should those people be thinking about the current era of proteomics to make the best possible decisions?
GARY
Yeah, probably the most important question — and I admit I’m entirely conflicted here, given the disclosures we’ve covered. I’ve participated in a lot of such decision making, most on the industrial side as well as the academic side. I think at the end of the day, first of all we’ve outlined that it’s very context dependent. The person leading this decision making should have a candid discussion with their team about what it is they want to get out of the analysis, and whether it is in fact targeted — a handful of proteins covered by affinity assays, or even a thousand proteins covered by affinity assays — that’s an informed decision one could make. But I think what I would recommend, because these technologies are all complicated and there is a lot of marketing associated with each of them, is this: these are important decisions, and I would encourage people to look at the actual data. There are a number of studies now that have come out rigorously comparing the technologies, and a number of ring trials that have looked at the pros and cons. And like I said, pros can be great for one study, but that pro may not matter for your specific context — coverage doesn’t matter if you only care about a thousand proteins. So I would encourage everyone to look at that hard data and try to use the actual results to make a decision, understanding the pros and cons. Every technology we talked about today has pros and cons. Just understand what they are so that whatever you move forward with, at least it’s an informed decision. It may end up being the wrong decision, but at least it was made based on deliberation.
DIPENDER
Sounds like democracy, Gary.