
by ODWAdmin | Sep 6, 2024
Improved Cell-Cultivated Seafood Production through Untargeted Metabolomics: A Study with BlueNalu® This case study with cell-cultivated seafood producer BlueNalu® showcases the power of Next-Generation Metabolomics® from Panome Bio to aid in the optimization of...
by ODWAdmin | Aug 16, 2024
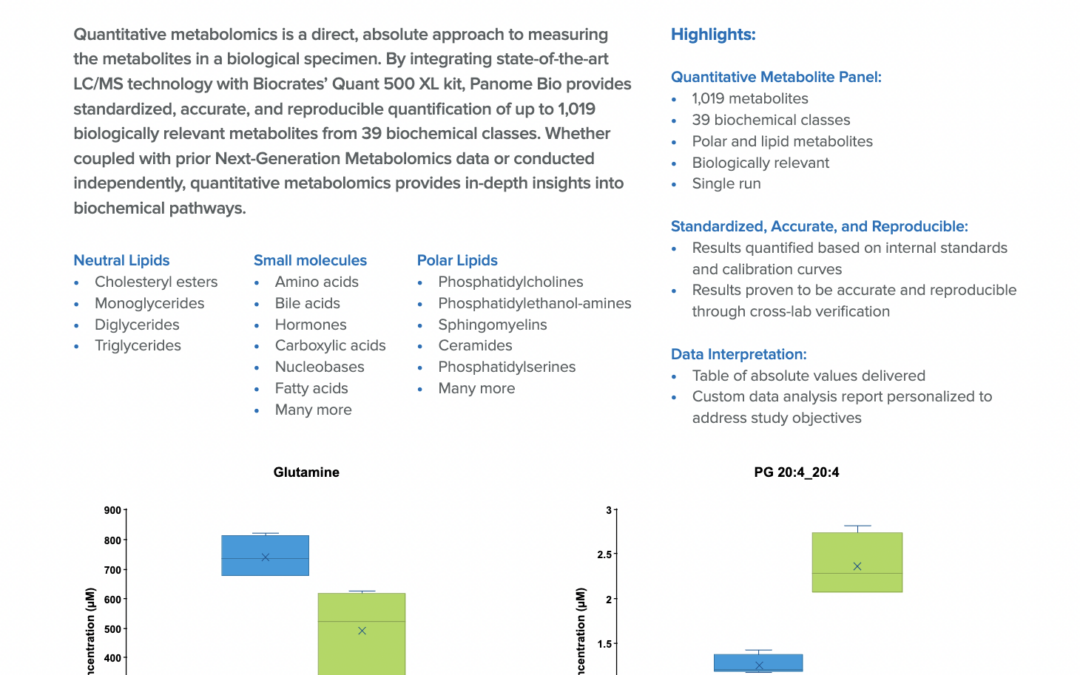
Targeted Metabolite Panels for Quantitative Measurements Explore the targeted, quantitative metabolite panel service offering from Panome Bio in this application note. Gain valuable insights into a biological system by quantifying metabolite and lipid...
| Protein Name | UniProt Accession Number |
|---|---|
| Adiponectin | Q60994 |
| Afamin | O89020 |
| Alpha-1-acid glycoprotein 2 | P07361 |
| Alpha-1-antitrypsin 1-1 | P07758 |
| Alpha-1-antitrypsin 1-4 | Q00897 |
| Alpha-1B-glycoprotein | Q19LI2 |
| Alpha-2-antiplasmin | Q61247 |
| Alpha-enolase | P17182 |
| Angiotensinogen | P11859 |
| Antithrombin-III | P32261 |
| Apolipoprotein A-II | P09813 |
| Apolipoprotein A-IV | P06728 |
| Apolipoprotein C-IV | Q61268 |
| Apolipoprotein D | P51910 |
| Apolipoprotein M | Q9Z1R3 |
| Bisphosphoglycerate mutase | P15327 |
| BPI fold-containing family A member 2 | P07743 |
| Cadherin-5 | P55284 |
| Carbonic anhydrase 2 | P00920 |
| Carboxypeptidase B2 | Q9JHH6 |
| Carboxypeptidase N catalytic chain | Q9JJN5 |
| Carboxypeptidase Q | Q9WVJ3 |
| Cartilage oligomeric matrix protein | Q9R0G6 |
| Cathepsin D | P18242 |
| CD5 antigen-like | Q9QWK4 |
| Ceruloplasmin | Q61147 |
| Coagulation factor V | O88783 |
| Coagulation factor X | O88947 |
| Coagulation factor XII | Q80YC5 |
| Coagulation factor XIII A chain | Q8BH61 |
| Coagulation factor XIII B chain | Q07968 |
| Collagen alpha-2(I) chain | Q01149 |
| Collectin-11 | Q3SXB8 |
| Complement C1q subcomponent subunit A | P98086 |
| Complement C1q subcomponent subunit C | Q02105 |
| Complement C1r subcomponent-like protein | Q3UZ09 |
| Complement C3 | P01027 |
| Complement C4-B | P01029 |
| Complement component C8 beta chain | Q8BH35 |
| Complement component C8 gamma chain | Q8VCG4 |
| Complement factor I | Q61129 |
| Corticosteroid-binding globulin | Q06770 |
| C-reactive protein | P14847 |
| Dermokine | Q6P253 |
| Dipeptidase 2 | Q8C255 |
| Endoplasmic reticulum chaperone BiP | P20029 |
| Endosialin | Q91V98 |
| Epidermal growth factor receptor | Q01279 |
| Extracellular matrix protein 1 | Q61508 |
| Extracellular superoxide dismutase | O09164 |
| Fetuin-B | Q9QXC1 |
| Fibrinogen beta chain | Q8K0E8 |
| Fibrinogen gamma chain | Q8VCM7 |
| Fibronectin | P11276 |
| Fructose-bisphosphate aldolase A | P05064 |
| Fructose-bisphosphate aldolase B | Q91Y97 |
| Gamma-glutamyl hydrolase | Q9Z0L8 |
| Gelsolin | P13020 |
| Glial fibrillary acidic protein | P03995 |
| Glutathione peroxidase 3 | P46412 |
| Glutathione S-transferase A3 | P30115 |
| H-2 class I histocompatibility antigen, Q10 alpha chain | P01898 |
| Haptoglobin | Q61646 |
| Heat shock protein HSP 90-beta | P11499 |
| Hemoglobin subunit beta-1 | P02088 |
| Hemopexin | Q91X72 |
| Heparin cofactor 2 | P49182 |
| Histidine-rich glycoprotein | Q9ESB3 |
| Histone H4 | P62806 |
| Hyaluronan-binding protein 2 | Q8K0D2 |
| Ig gamma-1 chain C region, membrane-bound form | P01869 |
| Ig gamma-2A chain C region secreted form | P01864 |
| Ig gamma-2B chain C region | P01867 |
| Immunoglobulin J chain | P01592 |
| Immunoglobulin kappa constant | P01837 |
| Inhibitor of carbonic anhydrase | Q9DBD0 |
| Inorganic pyrophosphatase | Q9D819 |
| Insulin-like growth factor-binding protein 3 | P47878 |
| Inter alpha-trypsin inhibitor, heavy chain 4 | A6X935 |
| Inter-alpha-trypsin inhibitor heavy chain H1 | Q61702 |
| Inter-alpha-trypsin inhibitor heavy chain H2 | Q61703 |
| Inter-alpha-trypsin inhibitor heavy chain H3 | Q61704 |
| Intercellular adhesion molecule 1 | P13597 |
| Intercellular adhesion molecule 2 | P35330 |
| Interleukin-1 receptor accessory protein | Q61730 |
| Interleukin-18-binding protein | Q9Z0M9 |
| Kininogen-1 | O08677 |
| Leukemia inhibitory factor receptor | P42703 |
| Lumican | P51885 |
| Macrophage colony-stimulating factor 1 receptor | P09581 |
| Mannan-binding lectin serine protease 2 | Q91WP0 |
| Metalloproteinase inhibitor 2 | P25785 |
| Microfibril-associated glycoprotein 4 | Q9D1H9 |
| N-acetylmuramoyl-L-alanine amidase | Q8VCS0 |
| Osteopontin | P10923 |
| Peroxiredoxin-1 | P35700 |
| Peroxiredoxin-2 | Q61171 |
| Phospholipid transfer protein | P55065 |
| Pigment epithelium-derived factor | P97298 |
| Plasma kallikrein | P26262 |
| Plasma protease C1 inhibitor | P97290 |
| Plasminogen | P20918 |
| Platelet factor 4 | Q9Z126 |
| Platelet glycoprotein Ib alpha chain | O35930 |
| Platelet glycoprotein V | O08742 |
| Proprotein convertase subtilisin/kexin type 9 | Q80W65 |
| Proteasome subunit alpha type-1 | Q9R1P4 |
| Proteasome subunit alpha type-4 | Q9R1P0 |
| Protein AMBP | Q07456 |
| Protein S100-A11 | P50543 |
| Protein S100-A9 | P31725 |
| Protein Z-dependent protease inhibitor | Q8R121 |
| Pyruvate kinase PKLR | P53657 |
| Serotransferrin | Q921I1 |
| Serum amyloid A-1 protein | P05366 |
| Serum paraoxonase/arylesterase 1 | P52430 |
| Sulfhydryl oxidase 1 | Q8BND5 |
| Tenascin | Q80YX1 |
| Uteroglobin | Q06318 |
| Vascular cell adhesion protein 1 | P29533 |
| Vasorin | Q9CZT5 |
| Vitamin D-binding protein | P21614 |
| Vitamin K-dependent protein C | P33587 |
| Vitronectin | P29788 |
| Zinc-alpha-2-glycoprotein | Q64726 |
| 26S proteasome non-ATPase regulatory subunit 9 | Q9CR00 |
| 40S ribosomal protein S2 | P25444 |
| Actin, aortic smooth muscle | P62737 |
| Alpha-2-macroglobulin receptor-associated protein | P55302 |
| Alpha-actinin-1 | Q7TPR4 |
| Alpha-amylase 1 | P00687 |
| Alpha-synuclein | O55042 |
| Apolipoprotein B-100 | E9Q414 |
| ATP synthase subunit d, mitochondrial | Q9DCX2 |
| Beta-1,4-galactosyltransferase 1 | P15535 |
| Beta-2-glycoprotein 1 | Q01339 |
| Beta-enolase | P21550 |
| Bleomycin hydrolase | Q8R016 |
| Cadherin-1 | P09803 |
| Carbonic anhydrase 3 | P16015 |
| Carboxylesterase 1C | P23953 |
| Carboxylesterase 3A | Q63880 |
| Cathepsin S | O70370 |
| Cathepsin Z | Q9WUU7 |
| Cell growth regulator with EF hand domain protein 1 | Q8R1U2 |
| Chitotriosidase-1 | Q9D7Q1 |
| Cholinesterase | Q03311 |
| Chromogranin-A | P26339 |
| Clusterin | Q06890 |
| Coagulation factor IX | P16294 |
| Coagulation factor VII | P70375 |
| Cofilin-1 | P18760 |
| Collagen alpha-1(I) chain | P11087 |
| Collagen alpha-1(III) chain | P08121 |
| Complement component C8 alpha chain | Q8K182 |
| Complement component C9 | P06683 |
| Complement factor H | P06909 |
| Core histone macro-H2A.2 | Q8CCK0 |
| Cystatin-C | P21460 |
| Cytochrome c, somatic | P62897 |
| Di-N-acetylchitobiase | Q8R242 |
| Dyslexia-associated protein KIAA0319-like protein | Q8K135 |
| Dystroglycan | Q62165 |
| EF-hand domain-containing protein D2 | Q9D8Y0 |
| EGF-containing fibulin-like extracellular matrix protein 1 | Q8BPB5 |
| Electron transfer flavoprotein subunit beta | Q9DCW4 |
| Elongation factor 1-alpha 1 | P10126 |
| Elongation factor 1-beta | O70251 |
| Endoplasmic reticulum aminopeptidase 1 | Q9EQH2 |
| Eukaryotic translation initiation factor 3 subunit E | P60229 |
| Ezrin | P26040 |
| Fibrinogen alpha chain | E9PV24 |
| Ficolin-1 | O70165 |
| Fumarylacetoacetase | P35505 |
| Galectin-1 | P16045 |
| Glucosidase 2 subunit beta | O08795 |
| Glutathione reductase, mitochondrial | P47791 |
| Glutathione S-transferase Mu 1 | P10649 |
| Glycerol-3-phosphate phosphatase | Q8CHP8 |
| Hemoglobin subunit alpha | P01942 |
| Hemoglobin subunit Zeta|Hemoglobin subunit alpha | P06467|P01942 |
| Hepatocyte growth factor activator | Q9R098 |
| Ig alpha chain C region | P01878 |
| Ig heavy chain V region 102 | P01750 |
| Ig heavy chain V region MOPC 47A | P01786 |
| Ig kappa chain V19-17 | P01633 |
| Ig kappa chain V-II region 7S34.1 | P01630 |
| Ig kappa chain V-III region PC 2154 | P01674 |
| Ig kappa chain V-V region MOPC 149 | P01636 |
| Ig kappa chain V-V region MOPC 173 | P01643 |
| Immunoglobulin heavy constant mu | P01872 |
| Insulin-like growth factor I | P05017 |
| Keratin, type I cytoskeletal 14 | Q61781 |
| Keratin, type I cytoskeletal 16 | Q9Z2K1 |
| Keratin, type II cytoskeletal 2 epidermal | Q3TTY5 |
| Keratin, type II cytoskeletal 5 | Q922U2 |
| Kunitz-type protease inhibitor 1 | Q9R097 |
| Lactoylglutathione lyase | Q9CPU0 |
| Low molecular weight phosphotyrosine protein phosphatase | Q9D358 |
| L-selectin | P18337 |
| Lysosomal alpha-mannosidase | O09159 |
| Mesothelin | Q61468 |
| Murinoglobulin-1 | P28665 |
| N(G),N(G)-dimethylarginine dimethylaminohydrolase 1 | Q9CWS0 |
| Neural cell adhesion molecule 1 | P13595 |
| Nucleobindin-1 | Q02819 |
| Nucleosome assembly protein 1-like 4 | Q78ZA7 |
| Out at first protein homolog | Q8QZR4 |
| Pancreatic alpha-amylase | P00688 |
| Papilin | Q9EPX2 |
| Perilipin-3 | Q9DBG5 |
| Peroxiredoxin-4 | O08807 |
| Peroxiredoxin-6 | O08709 |
| Platelet-activating factor acetylhydrolase | Q60963 |
| Plexin domain-containing protein 2 | Q9DC11 |
| Profilin-1 | P62962 |
| Progranulin | P28798 |
| Prolow-density lipoprotein receptor-related protein 1 | Q91ZX7 |
| Properdin | P11680 |
| Proteasome activator complex subunit 2 | P97372 |
| Proteasome subunit alpha type-5 | Q9Z2U1 |
| Proteasome subunit alpha type-6 | Q9QUM9 |
| Proteasome subunit beta type-1 | O09061 |
| Proteasome subunit beta type-4 | P99026 |
| Protein DEK | Q7TNV0 |
| Proteoglycan 4 | Q9JM99 |
| P-selectin | Q01102 |
| Pyruvate kinase PKM | P52480 |
| Radixin | P26043 |
| Serine protease inhibitor A3C | P29621 |
| Serine protease inhibitor A3M | Q03734 |
| Serine protease inhibitor A3N | Q91WP6 |
| Serum amyloid A-4 protein | P31532 |
| Sorbitol dehydrogenase | Q64442 |
| SPARC | P07214 |
| Spermine oxidase | Q99K82 |
| TBC1 domain family member 10A | P58802 |
| Thioredoxin | P10639 |
| Thrombospondin-1 | P35441 |
| Thyroxine-binding globulin | P61939 |
| Transaldolase | Q93092 |
| Transcobalamin-2 | O88968 |
| Transketolase | P40142 |
| Translocon-associated protein subunit alpha | Q9CY50 |
| Transthyretin | P07309 |
| Tropomyosin alpha-1 chain | P58771 |
| Ubiquitin-conjugating enzyme E2 N | P61089 |
| Ubiquitin-like protein ISG15 | Q64339 |
| Uromodulin | Q91X17 |
| WD repeat-containing protein 1 | O88342 |
| 14-3-3 protein sigma | O70456 |
| 26S proteasome regulatory subunit 6A | O88685 |
| 40S ribosomal protein S20 | P60867 |
| 60 kDa heat shock protein, mitochondrial | P63038 |
| 6-phosphogluconolactonase | Q9CQ60 |
| Actin, cytoplasmic 2 | P63260 |
| ADAMTS-like protein 4 | Q80T21 |
| Adipolin | Q8R2Z0 |
| Alpha-1-acid glycoprotein 1 | Q60590 |
| Alpha-1-antitrypsin 1-2 | P22599 |
| Alpha-1-antitrypsin 1-5 | Q00898 |
| Alpha-2-macroglobulin-P | Q6GQT1 |
| Alpha-hemoglobin-stabilizing protein | Q9CY02 |
| Aminoacyl tRNA synthase complex-interacting multifunctional protein 1 | P31230 |
| Angiogenin | P21570 |
| Angiopoietin-1 receptor | Q02858 |
| Apolipoprotein A-I | Q00623 |
| Apolipoprotein E | P08226 |
| Arginase-1 | Q61176 |
| ATP synthase subunit beta, mitochondrial | P56480 |
| Atrial natriuretic peptide receptor 1 | P18293 |
| Beta-2-microglobulin | P01887 |
| Bifunctional purine biosynthesis protein PURH | Q9CWJ9 |
| Calpastatin | P51125 |
| CapZ-interacting protein | Q3UZA1 |
| Carbonic anhydrase 1 | P13634 |
| Carboxylesterase 1D | Q8VCT4 |
| Carboxypeptidase N subunit 2 | Q9DBB9 |
| Cathepsin B | P10605 |
| Caveolae-associated protein 2 | Q63918 |
| CD97 antigen | Q9Z0M6 |
| Coactosin-like protein | Q9CQI6 |
| Coiled-coil domain-containing protein 25 | Q78PG9 |
| Collagen alpha-1(XII) chain | Q60847 |
| Collagen alpha-2(XI) chain | Q64739 |
| Complement C1q subcomponent subunit B | P14106 |
| Complement C1s-A subcomponent | Q8CG14 |
| Complement C2 | P21180 |
| Complement C5 | P06684 |
| Copper transport protein ATOX1 | O08997 |
| Coronin-1C | Q9WUM4 |
| Cysteine-rich protein 2 | Q9DCT8 |
| Cytosol aminopeptidase | Q9CPY7 |
| Deaminated glutathione amidase | Q8VDK1 |
| Desmoglein-2 | O55111 |
| Eukaryotic initiation factor 4A-I | P60843 |
| Eukaryotic translation initiation factor 3 subunit I | Q9QZD9 |
| Eukaryotic translation initiation factor 4B | Q8BGD9 |
| Exostosin-1 | P97464 |
| Ferritin heavy chain | P09528 |
| Fibrinogen-like protein 1 | Q71KU9 |
| Flavin reductase (NADPH) | Q923D2 |
| Follistatin-related protein 1 | Q62356 |
| Glutamate receptor ionotropic, NMDA 2A | P35436 |
| Glutamine synthetase | P15105 |
| Glycerol-3-phosphate dehydrogenase | P13707 |
| Glycogen phosphorylase, liver form | Q9ET01 |
| Glycosylation-dependent cell adhesion molecule 1 | Q02596 |
| Golgi-associated PDZ and coiled-coil motif-containing protein | Q8BH60 |
| Growth/differentiation factor 8 | O08689 |
| GTP-binding nuclear protein Ran | P62827 |
| Heme-binding protein 1 | Q9R257 |
| Ig gamma-3 chain C region | P03987 |
| Ig kappa chain V-II region 17S29.1 | P03976 |
| Ig kappa chain V-II region 26-10 | P01631 |
| Ig kappa chain V-IV region S107B | P01680 |
| Ig kappa chain V-V region K2 (Fragment) | P01635 |
| Ig kappa chain V-VI region NQ2-6.1 | P04945 |
| Insulin-like growth factor II | P09535 |
| Insulin-like growth factor-binding protein 6 | P47880 |
| Insulin-like growth factor-binding protein complex acid labile subunit | P70389 |
| Kappa-casein | P06796 |
| Keratin, type II cytoskeletal 2 oral | Q3UV17 |
| Keratin, type II cytoskeletal 8 | P11679 |
| Lactotransferrin | P08071 |
| Laminin subunit alpha-2 | Q60675 |
| Leptin receptor | P48356 |
| Liprin-alpha-2 | Q8BSS9 |
| L-lactate dehydrogenase B chain | P16125 |
| Lysosomal protective protein | P16675 |
| Lysosome-associated membrane glycoprotein 1 | P11438 |
| Macrophage migration inhibitory factor | P34884 |
| Mannan-binding lectin serine protease 1 | P98064 |
| Mannose-binding protein A | P39039 |
| Mannose-binding protein C | P41317 |
| Matrix metalloproteinase-17 | Q9R0S3 |
| MLV-related proviral Env polyprotein | P10404 |
| Myeloperoxidase | P11247 |
| Myosin light polypeptide 6 | Q60605 |
| Nidogen-1 | P10493 |
| Noelin | O88998 |
| Pappalysin-1 | Q8R4K8 |
| Peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 | Q9QUR7 |
| Phosphatidylcholine-sterol acyltransferase | P16301 |
| Phosphatidylethanolamine-binding protein 1 | P70296 |
| Phosphatidylinositol-glycan-specific phospholipase D | O70362 |
| Plastin-2 | Q61233 |
| Podocan | Q7TQ62 |
| Pregnancy zone protein | Q61838 |
| Prenylcysteine oxidase | Q9CQF9 |
| Procollagen C-endopeptidase enhancer 1 | Q61398 |
| Proline-rich acidic protein 1 | Q80XD8 |
| Proteasome subunit alpha type-2 | P49722 |
| Proteasome subunit beta type-10 | O35955 |
| Protein CTLA-2-alpha | P12399 |
| Protein disulfide-isomerase | P09103 |
| Protein phosphatase 1 regulatory subunit 7 | Q3UM45 |
| Prothrombin | P19221 |
| Retinoic acid receptor responder protein 2 | Q9DD06 |
| Retinol-binding protein 4 | Q00724 |
| Selenoprotein P | P70274 |
| Serum amyloid A-2 protein | P05367 |
| Serum amyloid P-component | P12246 |
| Serum paraoxonase/lactonase 3 | Q62087 |
| SPARC-like protein 1 | P70663 |
| Splicing factor 3B subunit 5 | Q923D4 |
| Superoxide dismutase [Cu-Zn] | P08228 |
| Thrombospondin-4 | Q9Z1T2 |
| Transforming growth factor beta-1 proprotein | P04202 |
| Tropomyosin alpha-3 chain | P21107-2 |
| Tyrosine-protein kinase receptor Tie-1 | Q06806 |
| UMP-CMP kinase | Q9DBP5 |
| Vasopressin-neurophysin 2-copeptin | P35455 |
| Vinculin | Q64727 |
| Vitamin K-dependent protein S | Q08761 |
| Protein Name | Gene names | UniProt Accession Number | Link | Peptide Sequence | Disease | Description |
|---|---|---|---|---|---|---|
| 60 kDa heat shock protein mitochondrial | HSPD1 HSP60 | P10809 | https://www.uniprot.org/uniprot/P10809 | GIIDPTK | spastic paraplegia 13 autosomal, autosomal dominant | neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. |
| N/L | N/L | N/L | N/L | N/L | leukodystrophy neurodegeneration | A severe autosomal recessive hypomyelinating leukodystrophy. Clinically characterized by infantile-onset rotary nystagmus, progressive spastic paraplegia, neurologic regression, motor impairment, profound mental retardation. |
| 72 KDa type IV collagenase | MMP2 CLG4A | P08253 | https://www.uniprot.org/uniprot/P08253 | IDAVYEAPQEEK | Multicentric osteolysis, nodulosis and arthopathy (MONA) | An autosomal recessive syndrome characterized by severe multicentric osteolysis with predominant involvement of the hands and feet. |
| Endoplasmic reticulum chaperone BiP | HSPA5 GRP78 | P11021 | https://www.uniprot.org/uniprot/P11021 | ITPSYVAFTPEGER | Autoantigen in rheumatoid arthritis. | N/L |
| A disintegrin and metalloproteinase with thrombospondin motifs 2 | ADAMTS2 PCINP PCPNI | O95450 | https://www.uniprot.org/uniprot/O95450 | IILLSYGK | Ehlers-Danlos syndrome, dermatosparaxis type (EDSDERMS) | A form of Ehlers-Danlos syndrome, a group of connective tissue disorders characterized by skin hyperextensibility, articular hypermobility, and tissue fragility. EDSDERMS is an autosomal recessive form characterized by extreme skin fragility and easy bruising, large fontanels, blue sclerae, puffy eyelids, micrognathia, umbilical hernia, and short fingers. |
| A disintegrin and metalloproteinase with thrombospondin motifs 20 | ADAMTS20 | P59510 | https://www.uniprot.org/uniprot/P59510 | IPAGATNVDIR | 0 | N/L |
| A disintegrin and metalloproteinase with thrombospondin motifs 9 | ADAMTS9 KIAA1312 | Q9P2N4 | https://www.uniprot.org/uniprot/Q9P2N4 | LYNPDVR | 0 | N/L |
| Actin, aortic smooth muscle | Actin, alpha cardiac muscle 1 | 8 other actin and actin-like proteins | ACTA2 ACTSA ACTVS GIG46 | P62736 | https://www.uniprot.org/uniprot/P62736 | SYELPDGQVITIGNER | ACTA2 mutations predispose patients to a variety of diffuse and diverse vascular diseases, premature onset coronary artery disease (CAD), premature ischemic strokes and Moyamoya disease. | N/L |
| N/L | N/L | N/L | N/L | https://www.uniprot.org/uniprot/P62736 | Aortic aneurysm, familial thoracic 6 (AAT6) | A disease characterized by permanent dilation of the thoracic aorta usually due to degenerative changes in the aortic wall. It is primarily associated with a characteristic histologic appearance known as ‘medial necrosis’ or ‘Erdheim cystic medial necrosis’ in which there is degeneration and fragmentation of elastic fibers, loss of smooth muscle cells, and an accumulation of basophilic ground substance. |
| N/L | N/L | N/L | N/L | N/L | Moyamoya disease 5 (MYMY5) | A progressive cerebral angiopathy characterized by bilateral intracranial carotid artery stenosis and telangiectatic vessels in the region of the basal ganglia. The abnormal vessels resemble a ‘puff of smoke’ (moyamoya) on cerebral angiogram. Affected individuals can develop transient ischemic attacks and/or cerebral infarction, and rupture of the collateral vessels can cause intracranial hemorrhage. Hemiplegia of sudden onset and epileptic seizures constitute the prevailing presentation in childhood, while subarachnoid bleeding occurs more frequently in adults. |
| N/L | N/L | N/L | N/L | N/L | Multisystemic smooth muscle dysfunction syndrome (MSMDYS) | A syndrome characterized by dysfunction of smooth muscle cells throughout the body, leading to aortic and cerebrovascular disease, fixed dilated pupils, hypotonic bladder, malrotation, and hypoperistalsis of the gut and pulmonary hypertension. |
| Actin, aortic smooth muscle | Actin, alpha cardiac muscle 1 | 8 other actin and actin-like proteins | ACTC1 ACTC | P68032 | https://www.uniprot.org/uniprot/P68032 | SYELPDGQVITIGNER | 1. Cardiomyopathy, dilated 1R (CMD1R) | A disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. |
| N/L | N/L | N/L | N/L | https://www.uniprot.org/uniprot/P68032 | Cardiomyopathy, familial hypertrophic 11 (CMH11) | A hereditary heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death. |
| N/L | N/L | N/L | N/L | N/L | Atrial septal defect 5 (ASD5}. | A congenital heart malformation characterized by incomplete closure of the wall between the atria resulting in blood flow from the left to the right atria. |
| Adhesion G protein-coupled receptor F5 | ADGRF5 GPR116 KIAA0758 | Q8IZF2 | https://www.uniprot.org/uniprot/Q8IZF2 | DVIVHPLPLK | 0 | N/L |
| Adipocyte plasma membrane-associated protein | APMAP C20orf3 UNQ1869/PRO4305 | Q9HDC9 | https://www.uniprot.org/uniprot/Q9HDC9 | LLEYDTVTR | 0 | N/L |
| Adiponectin | ADIPOQ ACDC ACRP30 APM1 GBP28 | Q15848 | https://www.uniprot.org/uniprot/Q15848 | IFYNQQNHYDGSTGK | Adiponectin deficiency (ADPND) | condition that results in very low concentrations of plasma adiponectin. |
| N/L | N/L | N/L | N/L | N/L | Diabetes mellitus, non-insulin-dependent (NIDDM) | A multifactorial disorder of glucose homeostasis caused by a lack of sensitivity to the body’s own insulin. Affected individuals usually have an obese body habitus and manifestations of a metabolic syndrome characterized by diabetes, insulin resistance, hypertension and hypertriglyceridemia. The disease results in long-term complications that affect the eyes, kidneys, nerves, and blood vessels. |
| ADM | ADM AM | P35318 | https://www.uniprot.org/uniprot/P35318 | LDVASEFR | 0 | N/L |
| Afamin | AFM ALB2 ALBA | P43652 | https://www.uniprot.org/uniprot/P43652 | DADPDTFFAK | 0 | N/L |
| Alpha-1-acid glycoprotein 1 | ORM1 AGP1 | P02763 | https://www.uniprot.org/uniprot/P02763 | NWGLSVYADKPETTK | 0 | N/L |
| Alpha-1-antichymotrypsin | SERPINA3 AACT GIG24 GIG25 | P01011 | https://www.uniprot.org/uniprot/P01011 | EIGELYLPK | 0 | N/L |
| Alpha-1-antitrypsin | SERPINA1 AAT PI PRO0684 PRO2209 | P01009 | https://www.uniprot.org/uniprot/P01009 | SVLGQLGITK | Alpha-1-antitrypsin deficiency (A1ATD) | A disorder whose most common manifestation is emphysema, which becomes evident by the third to fourth decade. A less common manifestation of the deficiency is liver disease, which occurs in children and adults, and may result in cirrhosis and liver failure |
| Alpha-1B-glycoprotein_VAR_018369 | A1BG | P04217 | https://www.uniprot.org/uniprot/P04217 | LETPDFQLFK | 0 | N/L |
| Alpha-2-antiplasmin | SERPINF2 AAP PLI | P08697 | https://www.uniprot.org/uniprot/P08697 | LGNQEPGGQTALK | Alpha-2-plasmin inhibitor deficiency | An autosomal recessive disorder resulting in severe hemorrhagic diathesis. |
| Alpha-2-HS-glycoprotein | AHSG FETUA PRO2743 | P02765 | https://www.uniprot.org/uniprot/P02765 | FSVVYAK | Alopecia-mental retardation syndrome 1 (APMR1) | A rare autosomal recessive form of alopecia. APMR1 patients show loss of hair on the scalp, absence of eyebrows, eyelashes, axillary and pubic hair, and mild to severe mental retardation. |
| Alpha-2-macroglobulin | A2M CPAMD5 FWP007 | P01023 | https://www.uniprot.org/uniprot/P01023 | AIGYLNTGYQR | 0 | N/L |
| Angiogenin | ANG RNASE5 | P03950 | https://www.uniprot.org/uniprot/P03950 | DINTFIHGNK | Amyotrophic lateral sclerosis 9 (ALS9) | A neurodegenerative disorder affecting upper motor neurons in the brain and lower motor neurons in the brain stem and spinal cord, resulting in fatal paralysis. Sensory abnormalities are absent. The pathologic hallmarks of the disease include pallor of the corticospinal tract due to loss of motor neurons, presence of ubiquitin-positive inclusions within surviving motor neurons, and deposition of pathologic aggregates. The etiology of amyotrophic lateral sclerosis is likely to be multifactorial, involving both genetic and environmental factors. The disease is inherited in 5-10% of the cases. |
| Angiopoietin-related protein 3 | ANGPTL3 ANGPT5 UNQ153/PRO179 | Q9Y5C1 | https://www.uniprot.org/uniprot/Q9Y5C1 | DLVFSTWDHK | Hypobetalipoproteinemia, familial, 2 (FHBL2) | A disorder of lipid metabolism characterized by less than 5th percentile age- and sex-specific levels of low density lipoproteins, and dietary fat malabsorption. Affected individuals present with combined hypolipidemia, consisting of extremely low plasma levels of LDL cholesterol, HDL cholesterol, and triglycerides. |
| N/L | N/L | N/L | N/L | N/L | May be involved in atherosclerosis. | Plasma levels are closely associated with arterial wall thickness. |
| N/L | N/L | N/L | N/L | N/L | May be involved in nephrotic syndrome | N/L |
| Angiotensinogen | AGT SERPINA8 | P01019 | https://www.uniprot.org/uniprot/P01019 | ALQDQLVLVAAK | Essential hypertension (EHT) | A condition in which blood pressure is consistently higher than normal with no identifiable cause |
| N/L | N/L | N/L | N/L | N/L | Renal tubular dysgenesis (RTD) | Autosomal recessive severe disorder of renal tubular development characterized by persistent fetal anuria and perinatal death, probably due to pulmonary hypoplasia from early-onset oligohydramnios (the Potter phenotype). |
| Antithrombin-III | SERPINC1 AT3 PRO0309 | P01008 | https://www.uniprot.org/uniprot/P01008 | DDLYVSDAFHK | DISEASE: Antithrombin III deficiency (AT3D) | An important risk factor for hereditary thrombophilia, a hemostatic disorder characterized by a tendency to recurrent thrombosis. Antithrombin-III deficiency is classified into 4 types. Type I: characterized by a 50% decrease in antigenic and functional levels. Type II: has defects affecting the thrombin-binding domain. Type III: alteration of the heparin-binding domain. Plasma AT-III antigen levels are normal in type II and III. Type IV: consists of miscellaneous group of unclassifiable mutations. |
| Apolipoprotein A-I | APOA1 | P02647 | https://www.uniprot.org/uniprot/P02647 | ATEHLSTLSEK | High density lipoprotein deficiency 2 (HDLD2) | Inherited as autosomal dominant trait. It is characterized by moderately low HDL cholesterol, predilection toward premature coronary artery disease (CAD) and a reduction in cellular cholesterol efflux. |
| N/L | N/L | N/L | N/L | N/L | High density lipoprotein deficiency 1 (HDLD1) | Recessive disorder characterized by absence of high density lipoprotein (HDL) cholesterol from plasma, accumulation of cholesteryl esters, premature coronary artery disease (CAD), hepatosplenomegaly, recurrent peripheral neuropathy and progressive muscle wasting and weakness |
| N/L | N/L | N/L | N/L | N/L | Amyloidosis 8 (AMYL8) | A form of hereditary generalized amyloidosis. Clinical features include extensive visceral amyloid deposits, renal amyloidosis resulting in nephrotic syndrome, arterial hypertension, hepatosplenomegaly, cholestasis, petechial skin rash. There is no involvement of the nervous system. |
| N/L | N/L | N/L | N/L | N/L | APOA1 mutations may be involved in the pathogenesis of amyloid polyneuropathy-nephropathy Iowa type, also known as amyloidosis van Allen type or familial amyloid polyneuropathy type III | N/L |
| Apolipoprotein A-II | APOA2 | P02652 | https://www.uniprot.org/uniprot/P02652 | SPELQAEAK | 0 | N/L |
| Apolipoprotein A-IV | APOA4 | P06727 | https://www.uniprot.org/uniprot/P06727 | LGEVNTYAGDLQK | 0 | N/L |
| Apolipoprotein B-100 | APOB | P04114 | https://www.uniprot.org/uniprot/P04114 | FPEVDVLTK | Hypobetalipoproteinemia, familial, 1 (FHBL1) | A disorder of lipid metabolism characterized by less than 5th percentile age- and sex-specific levels of low density lipoproteins, and dietary fat malabsorption. Clinical presentation may vary from no symptoms to severe gastrointestinal and neurological dysfunction similar to abetalipoproteinemia. Most cases of FHBL1 result from nonsense mutations in the APOB gene that lead to a premature stop codon, which generate prematurely truncated apo B protein products |
| N/L | N/L | N/L | N/L | N/L | Familial ligand-defective apolipoprotein B-100 | Dominantly inherited disorder of lipoprotein metabolism leading to hypercholesterolemia and increased proneness to coronary artery disease (CAD). The plasma cholesterol levels are dramatically elevated due to impaired clearance of LDL particles by defective APOB/E receptors. |
| N/L | N/L | N/L | N/L | N/L | Note=Defects in APOB associated with defects in other genes (polygenic) can contribute to hypocholesterolemia. | N/L |
| Apolipoprotein C-I | APOC1 | P02654 | https://www.uniprot.org/uniprot/P02654 | EWFSETFQK | 0 | N/L |
| Apolipoprotein C-II | APOC2 APC2 | P02655 | https://www.uniprot.org/uniprot/P02655 | TYLPAVDEK | Hyperlipoproteinemia 1B (HLPP1B) | Autosomal recessive trait characterized by hypertriglyceridemia, xanthomas, and increased risk of pancreatitis and early atherosclerosis |
| Apolipoprotein C-III | APOC3 | P02656 | https://www.uniprot.org/uniprot/P02656 | GWVTDGFSSLK | Hyperalphalipoproteinemia 2 (HALP2) | A condition characterized by high levels of high density lipoprotein (HDL) and increased HDL cholesterol levels.. |
| Apolipoprotein C-IV | APOC4 | P55056 | https://www.uniprot.org/uniprot/P55056 | ELLETVVNR | 0 | N/L |
| Apolipoprotein D | APOD | P05090 | https://www.uniprot.org/uniprot/P05090 | NILTSNNIDVK | 0 | N/L |
| Apolipoprotein E | APOE | P02649 | https://www.uniprot.org/uniprot/P02649 | LGPLVEQGR | Hyperlipoproteinemia 3 (HLPP3) . | A disorder characterized by the accumulation of intermediate-density lipoprotein particles (IDL or broad-beta-lipoprotein) rich in cholesterol. Clinical features include xanthomas, yellowish lipid deposits in the palmar crease, or less specific on tendons and on elbows. The disorder rarely manifests before the third decade in men. In women, it is usually expressed only after the menopause. The influence of APOE on lipid levels is often suggested to have major implications for the risk of coronary artery disease (CAD) |
| N/L | N/L | N/L | N/L | N/L | Alzheimer disease 2 | A late-onset form of Alzheimer disease. Alzheimer disease is a neurodegenerative disorder characterized by progressive dementia, loss of cognitive abilities, and deposition of fibrillar amyloid proteins as intraneuronal neurofibrillary tangles, extracellular amyloid plaques and vascular amyloid deposits. |
| N/L | N/L | N/L | N/L | N/L | Sea-blue histiocyte disease | Characterized by splenomegaly, mild thrombocytopenia and, in the bone marrow, numerous histiocytes containing cytoplasmic granules which stain bright blue with the usual hematologic stains. The syndrome is the consequence of an inherited metabolic defect analogous to Gaucher disease and other sphingolipidoses |
| N/L | N/L | N/L | N/L | N/L | Lipoprotein glomerulopathy (LPG) | Uncommon kidney disease characterized by proteinuria, progressive kidney failure, and distinctive lipoprotein thrombi in glomerular capillaries. |
| N/L | N/L | N/L | N/L | N/L | Familial hypercholesterolemia (FH) | A common autosomal dominant disorder characterized by elevated serum low-density lipoprotein (LDL) cholesterol levels, which result in excess deposition of cholesterol in tissues and leads to xanthelasma, xanthomas, accelerated atherosclerosis and increased risk of premature coronary heart disease |
| Apolipoprotein F | APOF | Q13790 | https://www.uniprot.org/uniprot/Q13790 | SGVQQLIQYYQDQK | 0 | N/L |
| Apolipoprotein L1 | APOL1 APOL | O14791 | https://www.uniprot.org/uniprot/O14791 | VAQELEEK | DISEASE: Focal segmental glomerulosclerosis 4 | A renal pathology defined by the presence of segmental sclerosis in glomeruli and resulting in proteinuria, reduced glomerular filtration rate and progressive decline in renal function. Renal insufficiency often progresses to end-stage renal disease, a highly morbid state requiring either dialysis therapy or kidney transplantation. |
| Apolipoprotein M | APOM G3A NG20 HSPC336 | O95445 | https://www.uniprot.org/uniprot/O95445 | AFLLTPR | 0 | N/L |
| Apolipoprotein(a) | LPA | P08519 | https://www.uniprot.org/uniprot/P08519 | GTYSTTVTGR | 0 | N/L |
| Aromatase | CYP19A1 ARO1 CYAR CYP19 | P11511 | https://www.uniprot.org/uniprot/P11511 | NMLEMIFTPR | Aromatase excess syndrome (AEXS) | An autosomal dominant disorder characterized by increased extraglandular aromatization of steroids that presents with heterosexual precocity in males and isosexual precocity in females. |
| N/L | N/L | N/L | N/L | N/L | Aromatase deficiency (AROD) | A rare disease in which fetal androgens are not converted into estrogens due to placental aromatase deficiency. Thus, pregnant women exhibit a hirsutism, which spontaneously resolves after post-partum. At birth, female babies present with pseudohermaphroditism due to virilization of extern genital organs. In adult females, manifestations include delay of puberty, breast hypoplasia and primary amenorrhoea with multicystic ovaries. |
| Atrial natriuretic peptide receptor 1 | NPR1 ANPRA | P16066 | https://www.uniprot.org/uniprot/P16066 | ITDYGLESFR | 0 | N/L |
| Attractin | ATRN KIAA0548 MGCA | O75882 | https://www.uniprot.org/uniprot/O75882 | SVNNVVVR | 0 | N/L |
| Autism susceptibility gene 2 protein | AUTS2 KIAA0442 | Q8WXX7 | https://www.uniprot.org/uniprot/Q8WXX7 | ALSLASSSGSDK | Mental retardation, autosomal dominant 26 (MRD26) | A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. Additional MRD26 features include autism, short stature, microcephaly, cerebral palsy, and facial dysmorphisms |
| B-cell scaffold protein with ankyrin repeats | BANK1 | Q8NDB2 | https://www.uniprot.org/uniprot/Q8NDB2 | LTIVHHPGGK | Systemic lupus erythematosus (SLE) | A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. |
| Beta-2-glycoprotein 1 | APOH B2G1 | P02749 | https://www.uniprot.org/uniprot/P02749 | ATVVYQGER | 0 | N/L |
| Beta-2-microglobulin | B2M CDABP0092 HDCMA22P | P61769 | https://www.uniprot.org/uniprot/P61769 | VNHVTLSQPK | Immunodeficiency 43 (IMD43) | A disorder characterized by marked reduction in serum concentrations of immunoglobulins and albumin, and hypoproteinemia due to hypercatabolism. Patients may suffer from recurrent respiratory tract infections and severe skin disease |
| N/L | N/L | N/L | N/L | N/L | DISEASE: Amyloidosis 8 (AMYL8) | A form of hereditary generalized amyloidosis. Clinical features include extensive visceral amyloid deposits, renal amyloidosis resulting in nephrotic syndrome, arterial hypertension, hepatosplenomegaly, cholestasis, petechial skin rash. There is no involvement of the nervous system. |
| Beta-Ala-His dipeptidase | CNDP1 CN1 CPGL2 UNQ1915/PRO4380 | Q96KN2 | https://www.uniprot.org/uniprot/Q96KN2 | ALEQDLPVNIK | 0 | N/L |
| Beta-nerve growth factor | NGF NGFB | P01138 | https://www.uniprot.org/uniprot/P01138 | TTATDIK | Neuropathy, hereditary sensory and autonomic, 5 (HSAN5) | A form of hereditary sensory and autonomic neuropathy, a genetically and clinically heterogeneous group of disorders characterized by degeneration of dorsal root and autonomic ganglion cells, and by sensory and/or autonomic abnormalities. HSAN5 patients manifest loss of pain perception and impaired temperature sensitivity, ulcers, and in some cases self-mutilation. The autonomic involvement is variable. |
| Biotinidase | BTD | P43251 | https://www.uniprot.org/uniprot/P43251 | SHLIIAQVAK | Biotinidase deficiency (BTD deficiency) | A juvenile form of multiple carboxylase deficiency, an autosomal recessive disorder of biotin metabolism, characterized by ketoacidosis, hyperammonemia, excretion of abnormal organic acid metabolites, and dermatitis. Biotinidase deficiency is characterized by seizures, hypotonia, skin rash, alopecia, ataxia, hearing loss, and optic atrophy. If untreated, symptoms usually become progressively worse, and coma and death may occur. |
| C-reactive protein | CRP PTX1 | P02741 | https://www.uniprot.org/uniprot/P02741 | AFVFPK | 0 | N/L |
| C4b-binding protein alpha chain | C4BPA C4BP | P04003 | https://www.uniprot.org/uniprot/P04003 | EDVYVVGTVLR | 0 | N/L |
| Cadherin-13 | CDH13 CDHH | P55290 | https://www.uniprot.org/uniprot/P55290 | INENTGSVSVTR | 0 | N/L |
| Cadherin-5 | CDH5 | P33151 | https://www.uniprot.org/uniprot/P33151 | ELDSTGTPTGK | 0 | N/L |
| Calcitonin | CALCA CALC1 | P01258 | https://www.uniprot.org/uniprot/P01258 | FHTFPQTAIGVGAPGK | 0 | N/L |
| Calcitonin gene-related peptide 1 | CALCA CALC1 | P06881 | https://www.uniprot.org/uniprot/P06881 | NNFVPTNVGSK | 0 | N/L |
| Calponin-1 | CNN1 | P51911 | https://www.uniprot.org/uniprot/P51911 | VNVGVK | 0 | N/L |
| Carbonic anhydrase 1 | CA1 | P00915 | https://www.uniprot.org/uniprot/P00915 | VLDALQAIK | 0 | N/L |
| Carboxypeptidase B2 | CPB2 | Q96IY4 | https://www.uniprot.org/uniprot/Q96IY4 | IAWHVIR | 0 | N/L |
| Carboxypeptidase N catalytic chain | CPN1 ACBP | P15169 | https://www.uniprot.org/uniprot/P15169 | SIPQVSPVR | DISEASE: Carboxypeptidase N deficiency (CPND) | Patients affected present some combination of angioedema or chronic urticaria, as well as hay fever or asthma, and have also slightly depressed serum carboxy peptidase N, suggestive of autosomal recessive inheritance of this disorder. |
| Carboxypeptidase N subunit 2 | CPN2 ACBP | P22792 | https://www.uniprot.org/uniprot/P22792 | AGGSWDLAVQER | 0 | N/L |
| Cartilage acidic protein 1 | CRTAC1 ASPIC1 CEP68 | Q9NQ79 | https://www.uniprot.org/uniprot/Q9NQ79 | GVASLFAGR | 0 | N/L |
| Cathelicidin antimicrobial peptide | CAMP CAP18 FALL39 HSD26 | P49913 | https://www.uniprot.org/uniprot/P49913 | AIDGINQR | 0 | N/L |
| Cation-independent mannose-6-phosphate receptor | IGF2R MPRI | P11717 | https://www.uniprot.org/uniprot/P11717 | GHQAFDVGQPR | 0 | N/L |
| CD40 ligand | CD40LG CD40L TNFSF5 TRAP | P29965 | https://www.uniprot.org/uniprot/P29965 | SQFEGFVK | Immunodeficiency with hyper-IgM, type 1 (HIGM1) | Immunoglobulin isotype switch defect characterized by elevated concentrations of serum IgM and decreased amounts of all other isotypes. Affected males present at an early age (usually within the first year of life) recurrent bacterial and opportunistic infections, including Pneumocystis carinii pneumonia and intractable diarrhea due to cryptosporidium infection. Despite substitution treatment with intravenous immunoglobulin, the overall prognosis is rather poor, with a death rate of about 10% before adolescence. |
| CD44 antigen | CD44 LHR MDU2 MDU3 MIC4 | P16070 | https://www.uniprot.org/uniprot/P16070 | YGFIEGHVVIPR | 0 | N/L |
| CD5 antigen-like | CD5L API6 UNQ203/PRO229 | O43866 | https://www.uniprot.org/uniprot/O43866 | LVGGLHR | 0 | N/L |
| Actin-depolymerizing factor | N/L | B7Z2X4 | https://www.uniprot.org/uniprot/B7Z2X4 | EGGQTAPASTR | NA | N/L |
| Ceruloplasmin | CP | P00450 | https://www.uniprot.org/uniprot/P00450 | IYHSHIDAPK | Aceruloplasminemia (ACERULOP) | An autosomal recessive disorder of iron metabolism characterized by iron accumulation in the brain as well as visceral organs. Clinical features consist of the triad of retinal degeneration, diabetes mellitus and neurological disturbances |
| N/L | N/L | N/L | N/L | N/L | Note=Ceruloplasmin levels are decreased in Wilson disease, in which copper cannot be incorporated into ceruloplasmin in liver because of defects in the copper-transporting ATPase 2. | N/L |
| Cholesteryl ester transfer protein | CETP | P11597 | https://www.uniprot.org/uniprot/P11597 | GVSLFDIINPEIITR | Hyperalphalipoproteinemia 1 (HALP1) | A condition characterized by high levels of high density lipoprotein (HDL) and increased HDL cholesterol levels. |
| Cholinesterase | BCHE CHE1 | P06276 | https://www.uniprot.org/uniprot/P06276 | YLTLNTESTR | Butyrylcholinesterase deficiency (BCHED) | An autosomal recessive metabolic condition characterized by increased sensitivity to certain anesthetic drugs, including the muscle relaxants succinylcholine or mivacurium. BCHED results in slower hydrolysis of these drugs and, consequently, a prolonged neuromuscular block, leading to apnea. The duration of the prolonged apnea varies significantly depending on the extent of the enzyme deficiency |
| Chromogranin-A | CHGA | P10645 | https://www.uniprot.org/uniprot/P10645 | ELQDLALQGAK | 0 | N/L |
| Claudin-5 | CLDN5 AWAL TMVCF | O00501 | https://www.uniprot.org/uniprot/O00501 | PDLSFPVK | 0 | N/L |
| Clusterin | CLU APOJ CLI KUB1 AAG4 | P10909 | https://www.uniprot.org/uniprot/P10909 | ELDESLQVAER | 0 | N/L |
| Coagulation factor IX | F9 | P00740 | https://www.uniprot.org/uniprot/P00740 | SALVLQYLR | Hemophilia B (HEMB) | An X-linked blood coagulation disorder characterized by a permanent tendency to hemorrhage, due to factor IX deficiency. It is phenotypically similar to hemophilia A, but patients present with fewer symptoms. Many patients are asymptomatic until the hemostatic system is stressed by surgery or trauma |
| N/L | N/L | N/L | N/L | N/L | Note=Mutations in position 43 (Oxford-3, San Dimas) and 46 (Cambridge) prevents cleavage of the propeptide. Mutation in position 93 (Alabama) probably fails to bind to cell membranes . Mutation in position 191 (Chapel-Hill) or in position 226 (Nagoya or Hilo) prevent cleavage of the activation peptide | N/L |
| N/L | N/L | N/L | N/L | N/L | Thrombophilia, X-linked, due to factor IX defect (THPH8) | A hemostatic disorder characterized by a tendency to thrombosis. |
| Coagulation factor V | F5 | P12259 | https://www.uniprot.org/uniprot/P12259 | AEVDDVIQVR | Factor V deficiency (FA5D) D | A blood coagulation disorder leading to a hemorrhagic diathesis known as parahemophilia. |
| N/L | N/L | N/L | N/L | N/L | Thrombophilia due to activated protein C resistance (THPH2) | A hemostatic disorder due to defective degradation of factor V by activated protein C. It is characterized by a poor anticoagulant response to activated protein C resulting in tendency to thrombosis. |
| N/L | N/L | N/L | N/L | N/L | Budd-Chiari syndrome (BDCHS) | A syndrome caused by obstruction of hepatic venous outflow involving either the hepatic veins or the terminal segment of the inferior vena cava. Obstructions are generally caused by thrombosis and lead to hepatic congestion and ischemic necrosis. Clinical manifestations observed in the majority of patients include hepatomegaly, right upper quadrant pain and abdominal ascites. Budd-Chiari syndrome is associated with a combination of disease states including primary myeloproliferative syndromes and thrombophilia due to factor V Leiden, protein C deficiency and antithrombin III deficiency. Budd-Chiari syndrome is a rare but typical complication in patients with polycythemia vera |
| N/L | N/L | N/L | N/L | N/L | Ischemic stroke (ISCHSTR) | A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors |
| N/L | N/L | N/L | N/L | N/L | Pregnancy loss, recurrent, 1 (RPRGL1) | A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. |
| Coagulation factor VII | F7 | P08709 | https://www.uniprot.org/uniprot/P08709 | VSQYIEWLQK | Factor VII deficiency (FA7D) | A hemorrhagic disease with variable presentation. The clinical picture can be very severe, with the early occurrence of intracerebral hemorrhages or repeated hemarthroses, or, in contrast, moderate with cutaneous-mucosal hemorrhages (epistaxis, menorrhagia) or hemorrhages provoked by a surgical intervention. Finally, numerous subjects are completely asymptomatic despite very low factor VII levels. |
| Coagulation factor VIII | F8 F8C | P00451 | https://www.uniprot.org/uniprot/P00451 | LHPTHYSIR | Hemophilia A (HEMA) | A disorder of blood coagulation characterized by a permanent tendency to hemorrhage |
| Coagulation factor X | F10 | P00742 | https://www.uniprot.org/uniprot/P00742 | MLEVPYVDR | Factor X deficiency (FA10D) | A hemorrhagic disease with variable presentation. Affected individuals can manifest prolonged nasal and mucosal hemorrhage, menorrhagia, hematuria, and occasionally hemarthrosis. Some patients do not have clinical bleeding diathesis. |
| Coagulation factor XI | F11 | P03951 | https://www.uniprot.org/uniprot/P03951 | TSESGLPSTR | Factor XI deficiency (FA11D) | A hemorrhagic disease characterized by reduced levels and activity of factor XI resulting in moderate bleeding symptoms, usually occurring after trauma or surgery. Patients usually do not present spontaneous bleeding but women can present with menorrhagia. Hemorrhages are usually moderate. |
| Coagulation factor XII | F12 | P00748 | https://www.uniprot.org/uniprot/P00748 | EQPPSLTR | Factor XII deficiency (FA12D) | An asymptomatic anomaly of in vitro blood coagulation. Its diagnosis is based on finding a low plasma activity of the factor in coagulating assays. It is usually only accidentally discovered through pre-operative blood tests. Factor XII deficiency is divided into two categories, a cross-reacting material (CRM)-negative group (negative F12 antigen detection) and a CRM-positive group (positive F12 antigen detection) |
| N/L | N/L | N/L | N/L | N/L | Hereditary angioedema 3 (HAE3) | An hereditary angioedema occurring only in women. Hereditary angioedema is an autosomal dominant disorder characterized by episodic local swelling involving subcutaneous or submucous tissue of the upper respiratory and gastrointestinal tracts, face, extremities, and genitalia. Hereditary angioedema type 3 differs from types 1 and 2 in that both concentration and function of C1 esterase inhibitor are normal. Hereditary angioedema type 3 is precipitated or worsened by high estrogen levels (e.g., during pregnancy or treatment with oral contraceptives) |
| Coagulation factor XIII A chain | F13A1 F13A | P00488 | https://www.uniprot.org/uniprot/P00488 | GTYIPVPIVSELQSGK | Factor XIII subunit A deficiency (FA13AD) | An autosomal recessive hematologic disorder characterized by a life-long bleeding tendency, impaired wound healing and spontaneous abortion in affected women. |
| Coagulation factor XIII B chain | F13B | P05160 | https://www.uniprot.org/uniprot/P05160 | IQTHSTTYR | Factor XIII subunit B deficiency (FA13BD) | An autosomal recessive hematologic disorder characterized by a life-long bleeding tendency, impaired wound healing and spontaneous abortion in affected women. |
| Collagen alpha-1(I) chain | COL1A1 | P02452 | https://www.uniprot.org/uniprot/P02452 | GVVGLPGQR | Caffey disease (CAFFD) | Caffey disease (CAFFD) [MIM:114000]: Characterized by an infantile episode of massive subperiosteal new bone formation that typically involves the diaphyses of the long bones, mandible, and clavicles. The involved bones may also appear inflamed, with painful swelling and systemic fever often accompanying the illness. The bone changes usually begin before 5 months of age and resolve before 2 years of age. {ECO:0000269|PubMed:15864348}. Note=The disease is caused by mutations affecting the gene represented in this entry.; |
| N/L | N/L | N/L | N/L | N/L | Ehlers-Danlos syndrome, classic type, 1 (EDSCL1) | A form of Ehlers-Danlos syndrome, a group of connective tissue disorders characterized by skin hyperextensibility, articular hypermobility, and tissue fragility. The main features of classic Ehlers-Danlos syndrome are joint hypermobility and dislocation, and fragile, bruisable skin. |
| N/L | N/L | N/L | N/L | N/L | Ehlers-Danlos syndrome, arthrochalasia type, 1 (EDSARTH1) | A form of Ehlers-Danlos syndrome, a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDSARTH1 is an autosomal dominant form characterized by frequent congenital hip dislocation and extreme joint laxity with recurrent joint subluxations and minimal skin involvement. |
| N/L | N/L | N/L | N/L | N/L | Osteogenesis imperfecta 1 (OI1) | An autosomal dominant form of osteogenesis imperfecta, a connective tissue disorder characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI1 is a non-deforming form with normal height or mild short stature, and no dentinogenesis imperfecta. |
| N/L | N/L | N/L | N/L | N/L | Osteogenesis imperfecta 2 (OI2) | An autosomal dominant form of osteogenesis imperfecta, a connective tissue disorder characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI2 is characterized by bone fragility, with many perinatal fractures, severe bowing of long bones, undermineralization, and death in the perinatal period due to respiratory insufficiency. |
| N/L | N/L | N/L | N/L | N/L | Osteogenesis imperfecta 3 (OI3) | An autosomal dominant form of osteogenesis imperfecta, a connective tissue disorder characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI3 is characterized by progressively deforming bones, very short stature, a triangular face, severe scoliosis, grayish sclera and dentinogenesis imperfecta |
| N/L | N/L | N/L | N/L | N/L | Osteogenesis imperfecta 4 (OI4) | An autosomal dominant form of osteogenesis imperfecta, a connective tissue disorder characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI4 is characterized by moderately short stature, mild to moderate scoliosis, grayish or white sclera and dentinogenesis imperfecta. |
| N/L | N/L | N/L | N/L | N/L | Osteoporosis (OSTEOP) | A systemic skeletal disorder characterized by decreased bone mass and deterioration of bone microarchitecture without alteration in the composition of bone. The result is fragile bones and an increased risk of fractures, even after minimal trauma. Osteoporosis is a chronic condition of multifactorial etiology and is usually clinically silent until a fracture occurs |
| Collagen alpha-1(III) chain | COL3A1 | P02461 | https://www.uniprot.org/uniprot/P02461 | GGAGPPGPEGGK | Ehlers-Danlos syndrome, vascular type (EDSVASC) | A severe form of Ehlers-Danlos syndrome, a group of connective tissue disorders characterized by skin hyperextensibility, articular hypermobility, and tissue fragility. EDSVASC is an autosomal dominant disease characterized by joint and dermal manifestations as in other forms of the syndrome, and by proneness to spontaneous rupture of bowel and large arteries. The vascular complications may affect all anatomical areas. |
| Collagen alpha-1(XVIII) chain | COL18A1 | P39060 | https://www.uniprot.org/uniprot/P39060 | AVGLAGTFR | Knobloch syndrome 1 (KNO1) | A developmental disorder primarily characterized by typical eye abnormalities, including high myopia, cataracts, dislocated lens, vitreoretinal degeneration, and retinal detachment, with occipital skull defects, which can range from occipital encephalocele to occult cutis aplasia. |
| Collagen alpha-2(I) chain | COL1A2 | P08123 | https://www.uniprot.org/uniprot/P08123 | GVVGPQGAR | Ehlers-Danlos syndrome, arthrochalasia type, 2 (EDSARTH2) | A form of Ehlers-Danlos syndrome, a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDSARTH2 is an autosomal dominant condition characterized by frequent congenital hip dislocation and extreme joint laxity with recurrent joint subluxations and minimal skin involvement. |
| N/L | N/L | N/L | N/L | N/L | Osteogenesis imperfecta 1 (OI1) | An autosomal dominant form of osteogenesis imperfecta, a connective tissue disorder characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI1 is a non-deforming form with normal height or mild short stature, and no dentinogenesis imperfecta. |
| N/L | N/L | N/L | N/L | N/L | Osteogenesis imperfecta 2 (OI2) | An autosomal dominant form of osteogenesis imperfecta, a connective tissue disorder characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI2 is characterized by bone fragility, with many perinatal fractures, severe bowing of long bones, undermineralization, and death in the perinatal period due to respiratory insufficiency. |
| N/L | N/L | N/L | N/L | N/L | Ehlers-Danlos syndrome, cardiac valvular type (EDSCV | A form of Ehlers-Danlos syndrome, a group of connective tissue disorders characterized by skin hyperextensibility, articular hypermobility, and tissue fragility. EDSCV is an autosomal recessive disease characterized by mitral valve prolapse and insufficiency, mitral regurgitation, and aortic insufficiency, in addition to joint laxity, skin hyperextensibility and friability, and abnormal scar formation. |
| N/L | N/L | N/L | N/L | N/L | Osteogenesis imperfecta 3 (OI3) | An autosomal dominant form of osteogenesis imperfecta, a connective tissue disorder characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI3 is characterized by progressively deforming bones, very short stature, a triangular face, severe scoliosis, grayish sclera and dentinogenesis imperfecta. |
| N/L | N/L | N/L | N/L | N/L | Osteogenesis imperfecta 4 (OI4) | An autosomal dominant form of osteogenesis imperfecta, a connective tissue disorder characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI4 is characterized by moderately short stature, mild to moderate scoliosis, grayish or white sclera and dentinogenesis imperfecta. |
| Complement C1q subcomponent subunit A | C1QA | P02745 | https://www.uniprot.org/uniprot/P02745 | PAFSAIR | Complement component C1q deficiency (C1QD) | A disorder caused by impaired activation of the complement classical pathway. It generally leads to severe immune complex disease with features of systemic lupus erythematosus and glomerulonephritis. Note=The disease is caused by mutations affecting the gene represented in this entry. |
| Complement C1q subcomponent subunit B | C1QB | P02746 | https://www.uniprot.org/uniprot/P02746 | IAFSATR | Complement component C1q deficiency (C1QD) | A disorder caused by impaired activation of the complement classical pathway. It generally leads to severe immune complex disease with features of systemic lupus erythematosus and glomerulonephritis. Note=The disease is caused by mutations affecting the gene represented in this entry. |
| Complement C1q subcomponent subunit C | C1QC C1QG | P02747 | https://www.uniprot.org/uniprot/P02747 | FQSVFTVTR | Complement component C1q deficiency (C1QD) | A disorder caused by impaired activation of the complement classical pathway. It generally leads to severe immune complex disease with features of systemic lupus erythematosus and glomerulonephritis. Note=The disease is caused by mutations affecting the gene represented in this entry. |
| Complement C1r subcomponent | C1R | P00736 | https://www.uniprot.org/uniprot/P00736 | GLTLHLK | Ehlers-Danlos syndrome, periodontal type, 1 (EDSPD1) | A form of Ehlers-Danlos syndrome, a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDSPD1 is characterized by the association of typical features of Ehlers-Danlos syndrome with gingival recession and severe early-onset periodontal disease, leading to premature loss of permanent teeth. EDSPD1 inheritance is autosomal dominant. |
| Complement C1r subcomponent-like protein | C1RL C1RL1 C1RLP CLSPA | Q9NZP8 | https://www.uniprot.org/uniprot/Q9NZP8 | VVVHPDYR | 0 | N/L |
| Complement C1s subcomponent | C1S | P09871 | https://www.uniprot.org/uniprot/P09871 | TNFDNDIALVR | Complement component C1s deficiency (C1SD) | A rare defect resulting in C1 deficiency and impaired activation of the complement classical pathway. C1 deficiency generally leads to severe immune complex disease with features of systemic lupus erythematosus and glomerulonephritis. |
| N/L | N/L | N/L | N/L | N/L | Ehlers-Danlos syndrome, periodontal type, 2 (EDSPD2) | A form of Ehlers-Danlos syndrome, a connective tissue disorder characterized by hyperextensible skin, atrophic cutaneous scars due to tissue fragility and joint hyperlaxity. EDSPD2 is characterized by the association of typical features of Ehlers-Danlos syndrome with gingival recession and severe early-onset periodontal disease, leading to premature loss of permanent teeth. EDSPD2 transmission pattern is consistent with autosomal dominant inheritance. |
| Complement C2 | C2 | P06681 | https://www.uniprot.org/uniprot/P06681 | HAFILQDTK | Complement component 2 deficiency (C2D) | A rare defect of the complement classical pathway associated with the development of autoimmune disorders, mainly systemic lupus erythematosus. Skin and joint manifestations are common and renal disease is relatively rare. Patients with complement component 2 deficiency are also reported to have recurrent invasive infections. |
| Complement C3 | C3 CPAMD1 | P01024 | https://www.uniprot.org/uniprot/P01024 | TGLQEVEVK | Complement component 3 deficiency (C3D) | A rare defect of the complement classical pathway. Patients develop recurrent, severe, pyogenic infections because of ineffective opsonization of pathogens. Some patients may also develop autoimmune disorders, such as arthralgia and vasculitic rashes, lupus-like syndrome and membranoproliferative glomerulonephritis. ; DISEASE: |
| N/L | N/L | N/L | N/L | N/L | Macular degeneration, age-related, 9 (ARMD9) | A form of age-related macular degeneration, a multifactorial eye disease and the most common cause of irreversible vision loss in the developed world. In most patients, the disease is manifest as ophthalmoscopically visible yellowish accumulations of protein and lipid that lie beneath the retinal pigment epithelium and within an elastin-containing structure known as Bruch membrane. |
| N/L | N/L | N/L | N/L | N/L | Hemolytic uremic syndrome atypical 5 (AHUS5) | An atypical form of hemolytic uremic syndrome. It is a complex genetic disease characterized by microangiopathic hemolytic anemia, thrombocytopenia, renal failure and absence of episodes of enterocolitis and diarrhea. In contrast to typical hemolytic uremic syndrome, atypical forms have a poorer prognosis, with higher death rates and frequent progression to end-stage renal disease. |
| N/L | N/L | N/L | N/L | N/L | Note=Increased levels of C3 and its cleavage product ASP, are associated with obesity, diabetes and coronary heart disease. Short-term endurance training reduces baseline ASP levels and subsequently fat storage. | N/L |
| Complement C5 | C5 CPAMD4 | P01031 | https://www.uniprot.org/uniprot/P01031 | VFQFLEK | Complement component 5 deficiency (C5D) | : A rare defect of the complement classical pathway associated with susceptibility to severe recurrent infections, predominantly by Neisseria gonorrhoeae or Neisseria meningitidis |
| N/L | N/L | N/L | N/L | N/L | Note=An association study of C5 haplotypes and genotypes in individuals with chronic hepatitis C virus infection shows that individuals homozygous for the C5_1 haplotype have a significantly higher stage of liver fibrosis than individuals carrying at least 1 other allele | N/L |
| Complement component C6 | C6 | P13671 | https://www.uniprot.org/uniprot/P13671 | DLHLSDVFLK | Complement component 6 deficiency (C6D | A rare defect of the complement classical pathway associated with susceptibility to severe recurrent infections, predominantly by Neisseria gonorrhoeae or Neisseria meningitidis. |
| Complement component C7 | C7 | P10643 | https://www.uniprot.org/uniprot/P10643 | AASGTQNNVLR | Complement component 7 deficiency (C7D) | A rare defect of the complement classical pathway associated with susceptibility to severe recurrent infections, predominantly by Neisseria gonorrhoeae or Neisseria meningitidis. |
| Complement component C8 alpha chain | C8A | P07357 | https://www.uniprot.org/uniprot/P07357 | MESLGITSR | Complement component 8 deficiency, 1 (C8D1) | A rare defect of the complement classical pathway associated with susceptibility to severe recurrent infections, predominantly by Neisseria gonorrhoeae or Neisseria meningitidis. |
| Complement component C8 beta chain | C8B | P07358 | https://www.uniprot.org/uniprot/P07358 | SDLEVAHYK | Complement component 8 deficiency, 2 (C8D2) | A rare defect of the complement classical pathway associated with susceptibility to severe recurrent infections, predominantly by Neisseria gonorrhoeae or Neisseria meningitidis. |
| Complement component C9 | C9 | P02748 | https://www.uniprot.org/uniprot/P02748 | LSPIYNLVPVK | Complement component 9 deficiency (C9D) | A rare defect of the complement classical pathway associated with susceptibility to severe recurrent infections predominantly by Neisseria gonorrhoeae or Neisseria meningitidis. Some patients may develop dermatomyositis. |
| N/L | N/L | N/L | N/L | N/L | Macular degeneration, age-related, 15 (ARMD15) | A form of age-related macular degeneration, a multifactorial eye disease and the most common cause of irreversible vision loss in the developed world. In most patients, the disease is manifest as ophthalmoscopically visible yellowish accumulations of protein and lipid that lie beneath the retinal pigment epithelium and within an elastin-containing structure known as Bruch membrane. |
| Complement factor B | CFB BF BFD | P00751 | https://www.uniprot.org/uniprot/P00751 | EELLPAQDIK | Hemolytic uremic syndrome atypical 4 (AHUS4) | An atypical form of hemolytic uremic syndrome. It is a complex genetic disease characterized by microangiopathic hemolytic anemia, thrombocytopenia, renal failure and absence of episodes of enterocolitis and diarrhea. In contrast to typical hemolytic uremic syndrome, atypical forms have a poorer prognosis, with higher death rates and frequent progression to end-stage renal disease. |
| N/L | N/L | N/L | N/L | N/L | Complement factor B deficiency (CFBD) | An immunologic disorder characterized by increased susceptibility to bacterial infections, particularly Neisseria infections, due to a defect in the alternative complement pathway. |
| Complement factor D | CFD DF PFD | P00746 | https://www.uniprot.org/uniprot/P00746 | THHDGAITER | Complement factor D deficiency (CFDD) | An immunologic disorder characterized by increased susceptibility to bacterial infections, particularly Neisseria infections, due to a defect in the alternative complement pathway. { |
| Complement factor H | CFH HF HF1 HF2 | P08603 | https://www.uniprot.org/uniprot/P08603 | SSQESYAHGTK | Basal laminar drusen (BLD) | DDrusen are extracellular deposits that accumulate below the retinal pigment epithelium on Bruch membrane. Basal laminar drusen refers to an early adult-onset drusen phenotype that shows a pattern of uniform small, slightly raised yellow subretinal nodules randomly scattered in the macula. In later stages, these drusen often become more numerous, with clustered groups of drusen scattered throughout the retina. In time these small basal laminar drusen may expand and ultimately lead to a serous pigment epithelial detachment of the macula that may result in vision loss. |
| N/L | N/L | N/L | N/L | N/L | Complement factor H deficiency (CFHD) | A disorder that can manifest as several different phenotypes, including asymptomatic, recurrent bacterial infections, and renal failure. Laboratory features usually include decreased serum levels of factor H, complement component C3, and a decrease in other terminal complement components, indicating activation of the alternative complement pathway. It is associated with a number of renal diseases with variable clinical presentation and progression, including membranoproliferative glomerulonephritis and atypical hemolytic uremic syndrome. |
| N/L | N/L | N/L | N/L | N/L | Hemolytic uremic syndrome atypical 1 (AHUS1) | An atypical form of hemolytic uremic syndrome. It is a complex genetic disease characterized by microangiopathic hemolytic anemia, thrombocytopenia, renal failure and absence of episodes of enterocolitis and diarrhea. In contrast to typical hemolytic uremic syndrome, atypical forms have a poorer prognosis, with higher death rates and frequent progression to end-stage renal disease. |
| N/L | N/L | N/L | N/L | N/L | Macular degeneration, age-related, 4 (ARMD4) | A form of age-related macular degeneration, a multifactorial eye disease and the most common cause of irreversible vision loss in the developed world. In most patients, the disease is manifest as ophthalmoscopically visible yellowish accumulations of protein and lipid that lie beneath the retinal pigment epithelium and within an elastin-containing structure known as Bruch membrane. |
| Complement factor I | CFI IF | P05156 | https://www.uniprot.org/uniprot/P05156 | VFSLQWGEVK | Hemolytic uremic syndrome atypical 3 (AHUS3) | An atypical form of hemolytic uremic syndrome. It is a complex genetic disease characterized by microangiopathic hemolytic anemia, thrombocytopenia, renal failure and absence of episodes of enterocolitis and diarrhea. In contrast to typical hemolytic uremic syndrome, atypical forms have a poorer prognosis, with higher death rates and frequent progression to end-stage renal disease.. |
| N/L | N/L | N/L | N/L | N/L | Complement factor I deficiency (CFI deficiency) | Autosomal recessive condition associated with a propensity to pyogenic infections. ; |
| N/L | N/L | N/L | N/L | N/L | Macular degeneration, age-related, 13 (ARMD13) | A form of age-related macular degeneration, a multifactorial eye disease and the most common cause of irreversible vision loss in the developed world. In most patients, the disease is manifest as ophthalmoscopically visible yellowish accumulations of protein and lipid that lie beneath the retinal pigment epithelium and within an elastin-containing structure known as Bruch membrane |
| Complement C4-A | C4A CO4 CPAMD2 | P0C0L4 | https://www.uniprot.org/uniprot/P0C0L4 | VLSLAQEQVGGSPEK | Complement component 4A deficiency (C4AD) | A rare defect of the complement classical pathway associated with the development of autoimmune disorders, mainly systemic lupus with or without associated glomerulonephritis |
| N/L | N/L | N/L | N/L | N/L | Systemic lupus erythematosus (SLE) | A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. The disease is marked by a wide range of system dysfunctions, an elevated erythrocyte sedimentation rate, and the formation of LE cells in the blood or bone marrow. |
| N/L | N/L | N/L | N/L | N/L | N/L | Note=Disease susceptibility is associated with variations affecting the gene represented in this entry. Interindividual copy-number variation (CNV) of complement component C4 and associated polymorphisms result in different susceptibilities to SLE. The risk of SLE susceptibility has been shown to be significantly increased among subjects with only two copies of total C4. A high copy number is a protective factor against SLE. |
| Complement C4-B | C4B CO4 CPAMD3; C4B_2 | P0C0L5 | https://www.uniprot.org/uniprot/P0C0L5 | N/L | Systemic lupus erythematosus (SLE) | A chronic, relapsing, inflammatory, and often febrile multisystemic disorder of connective tissue, characterized principally by involvement of the skin, joints, kidneys and serosal membranes. It is of unknown etiology, but is thought to represent a failure of the regulatory mechanisms of the autoimmune system. The disease is marked by a wide range of system dysfunctions, an elevated erythrocyte sedimentation rate, and the formation of LE cells in the blood or bone marrow. |
| N/L | N/L | N/L | N/L | N/L | Complement component 4B deficiency (C4BD) | A rare defect of the complement classical pathway associated with the development of autoimmune disorders, mainly systemic lupus with or without associated glomerulonephrit |
| Corticosteroid-binding globulin | SERPINA6 CBG | P08185 | https://www.uniprot.org/uniprot/P08185 | WSAGLTSSQVDLYIPK | Corticosteroid-binding globulin deficiency (CBG deficiency) | Extremely rare hereditary disorder characterized by reduced corticosteroid-binding capacity with normal or low plasma corticosteroid-binding globulin concentration, and normal or low basal cortisol levels associated with hypo/hypertension and muscle fatigue |
| Creatine kinase B-type | CKB CKBB | P12277 | https://www.uniprot.org/uniprot/P12277 | DLFDPIIEDR | 0 | N/L |
| Creatine kinase M-type | CKM CKMM | P06732 | https://www.uniprot.org/uniprot/P06732 | FEEILTR | 0 | N/L |
| Cystatin-C | CST3 | P01034 | https://www.uniprot.org/uniprot/P01034 | ALDFAVGEYNK | DISEASE: Amyloidosis 6 (AMYL6) | A hereditary generalized amyloidosis due to cystatin C amyloid deposition. Cystatin C amyloid accumulates in the walls of arteries, arterioles, and sometimes capillaries and veins of the brain, and in various organs including lymphoid tissue, spleen, salivary glands, and seminal vesicles. Amyloid deposition in the cerebral vessels results in cerebral amyloid angiopathy, cerebral hemorrhage and premature stroke. Cystatin C levels in the cerebrospinal fluid are abnormally low. |
| N/L | N/L | N/L | N/L | N/L | Macular degeneration, age-related, 11 (ARMD11) | A form of age-related macular degeneration, a multifactorial eye disease and the most common cause of irreversible vision loss in the developed world. In most patients, the disease is manifest as ophthalmoscopically visible yellowish accumulations of protein and lipid that lie beneath the retinal pigment epithelium and within an elastin-containing structure known as Bruch membrane |
| Desmoplakin | DSP | P15924 | https://www.uniprot.org/uniprot/P15924 | AELIVQPELK | Keratoderma, palmoplantar, striate 2 (SPPK2) [MIM:612908] | : A dermatological disorder characterized by thickening of the skin on the palms (linear pattern) and the soles (island-like pattern) and flexor aspect of the fingers. Abnormalities of the nails, the teeth and the hair are rarely present. |
| N/L | N/L | N/L | N/L | N/L | Cardiomyopathy, dilated, with woolly hair and keratoderma (DCWHK) | An autosomal recessive cardiocutaneous syndrome characterized by a generalized striate keratoderma particularly affecting the palmoplantar epidermis, woolly hair, and dilated left ventricular cardiomyopathy. |
| N/L | N/L | N/L | N/L | N/L | Arrhythmogenic right ventricular dysplasia, familial, 8 (ARVD8) | A congenital heart disease characterized by infiltration of adipose and fibrous tissue into the right ventricle and loss of myocardial cells, resulting in ventricular and supraventricular arrhythmias. |
| N/L | N/L | N/L | N/L | N/L | Skin fragility-woolly hair syndrome (SFWHS) | An autosomal recessive genodermatosis characterized by skin fragility with blistering, focal and diffuse palmoplantar keratoderma, hyperkeratotic plaques on the trunk and limbs, and woolly hair with varying degrees of alopecia |
| N/L | N/L | N/L | N/L | N/L | Epidermolysis bullosa, lethal acantholytic (EBLA) | A form of epidermolysis bullosa characterized by severe fragility of skin and mucous membranes. The phenotype is lethal in the neonatal period because of immense transcutaneous fluid loss. Typical features include universal alopecia, neonatal teeth, and nail loss. Histopathology of the skin shows suprabasal clefting and acantholysis throughout the spinous layer, mimicking pemphigus. |
| N/L | N/L | N/L | N/L | N/L | Cardiomyopathy, dilated, with woolly hair, keratoderma, and tooth agenesis (DCWHKTA) | A cardiocutaneous syndrome characterized by biventricular dilated cardiomyopathy, hyperkeratosis, woolly hair, palmoplantar keratoderma, and hypo/oligodontia. |
| Di-N-acetylchitobiase | CTBS CTB | Q01459 | https://www.uniprot.org/uniprot/Q01459 | ATYIQNYR | 0 | N/L |
| Dickkopf-related protein 1 and 2 | DKK1 | DKK2 | O94907|Q9UBU2 | https://www.uniprot.org/uniprot/O94907|Q9UBU2 | GSHGLEIFQR | 0 | N/L |
| E-selectin | SELE ELAM1 | P16581 | https://www.uniprot.org/uniprot/P16581 | YTHLVAIQNK | 0 | N/L |
| Elastin | ELN | P15502 | https://www.uniprot.org/uniprot/P15502 | LPGGYGLPYTTGK | Cutis laxa, autosomal dominant, 1 (ADCL1) | A connective tissue disorder characterized by loose, hyperextensible skin with decreased resilience and elasticity leading to a premature aged appearance. Face, hands, feet, joints, and torso may be differentially affected. Additional variable clinical features are gastrointestinal diverticula, hernia, and genital prolapse. Rare manifestations are pulmonary artery stenosis, aortic aneurysm, bronchiectasis, and emphysema. |
| N/L | N/L | N/L | N/L | N/L | Supravalvular aortic stenosis (SVAS) | Congenital narrowing of the ascending aorta which can occur sporadically, as an autosomal dominant condition, or as one component of Williams-Beuren syndrome |
| N/L | N/L | N/L | N/L | N/L | N/L | Note=ELN is located in the Williams-Beuren syndrome (WBS) critical region. WBS results from a hemizygous deletion of several genes on chromosome 7q11.23, thought to arise as a consequence of unequal crossing over between highly homologous low-copy repeat sequences flanking the deleted region. Haploinsufficiency of ELN may be the cause of certain cardiovascular and musculo-skeletal abnormalities observed in the disease |
| Endothelial lipase | LIPG UNQ387/PRO719 | Q9Y5X9 | https://www.uniprot.org/uniprot/Q9Y5X9 | LVSALHTR | 0 | N/L |
| Endothelial protein C receptor | PROCR EPCR | Q9UNN8 | https://www.uniprot.org/uniprot/Q9UNN8 | TLAFPLTIR | 0 | N/L |
| Epidermal growth factor receptor | EGFR ERBB ERBB1 HER1 | P00533 | https://www.uniprot.org/uniprot/P00533 | IPLENLQIIR | Lung cancer (LNCR) | A common malignancy affecting tissues of the lung. The most common form of lung cancer is non-small cell lung cancer (NSCLC) that can be divided into 3 major histologic subtypes: squamous cell carcinoma, adenocarcinoma, and large cell lung cancer. NSCLC is often diagnosed at an advanced stage and has a poor prognosis |
| N/L | N/L | N/L | N/L | N/L | Inflammatory skin and bowel disease, neonatal, 2 (NISBD2) | A disorder characterized by inflammatory features with neonatal onset, involving the skin, hair, and gut. The skin lesions involve perioral and perianal erythema, psoriasiform erythroderma, with flares of erythema, scaling, and widespread pustules. Gastrointestinal symptoms include malabsorptive diarrhea that is exacerbated by intercurrent gastrointestinal infections. The hair is short or broken, and the eyelashes and eyebrows are wiry and disorganized. |
| Extracellular matrix protein 1 | ECM1 | Q16610 | https://www.uniprot.org/uniprot/Q16610 | NVALVSGDTENAK | Lipoid proteinosis (LiP) | Rare autosomal recessive disorder characterized by generalized thickening of skin, mucosae and certain viscera. Classical features include beaded eyelid papules and laryngeal infiltration leading to hoarseness. Histologically, there is widespread deposition of hyaline material and disruption/reduplication of basement membrane. |
| Fatty acid-binding protein heart | FABP3 FABP11 MDGI | P05413 | https://www.uniprot.org/uniprot/P05413 | SLGVGFATR | 0 | N/L |
| Ferritin heavy chain | FTH1 FTH FTHL6 OK/SW-cl.84 PIG15 | P02794 | https://www.uniprot.org/uniprot/P02794 | NVNQSLLELHK | Hemochromatosis 5 (HFE5) | A disorder of iron metabolism characterized by iron overload. Excess iron is deposited in a variety of organs leading to their failure, and resulting in serious illnesses including cirrhosis, hepatomas, diabetes, cardiomyopathy, arthritis, and hypogonadotropic hypogonadism. Severe effects of the disease usually do not appear until after decades of progressive iron loading. Note=The disease is caused by mutations affecting the gene represented in this entry. In a Japanese family affected by HFE5, a single point mutation has been detected in the iron-responsive element (IRE) in the 5′-UTR of FTH1 mRNA. This mutation leads to an increased binding affinity for iron regulatory protein and thereby to the efficient suppression of mRNA translation. |
| Ferritin light chain | FTL | P02792 | https://www.uniprot.org/uniprot/P02792 | LGGPEAGLGEYLFER | Hyperferritinemia with or without cataract (HRFTC) | An autosomal dominant disease characterized by elevated level of ferritin in serum and tissues, and early-onset bilateral cataract. Cataracts may be subclinical in some patients. ; |
| N/L | N/L | N/L | N/L | N/L | Neurodegeneration with brain iron accumulation 3 (NBIA3) | A neurodegenerative disorder associated with iron accumulation in the brain, primarily in the basal ganglia. It is characterized by a variety of neurological signs including parkinsonism, ataxia, corticospinal signs, mild non-progressive cognitive deficit and episodic psychosis. It is linked with decreased serum ferritin levels. |
| N/L | N/L | N/L | N/L | N/L | L-ferritin deficiency (LFTD) | A condition characterized by low levels of ferritin in serum and tissues in the absence of other hematological symptoms. Seizures and mild neuropsychologic impairment may manifest in individuals with complete ferritin deficiency. |
| Fetuin-B | FETUB | Q9UGM5 | https://www.uniprot.org/uniprot/Q9UGM5 | LVVLPFPK | 0 | N/L |
| Fibrinogen alpha chain | FGA | P02671 | https://www.uniprot.org/uniprot/P02671 | VQHIQLLQK | Congenital afibrinogenemia (CAFBN) | Rare autosomal recessive disorder is characterized by bleeding that varies from mild to severe and by complete absence or extremely low levels of plasma and platelet fibrinogen. |
| N/L | N/L | N/L | N/L | N/L | Amyloidosis 8 (AMYL8) | A form of hereditary generalized amyloidosis. Clinical features include extensive visceral amyloid deposits, renal amyloidosis resulting in nephrotic syndrome, arterial hypertension, hepatosplenomegaly, cholestasis, petechial skin rash. There is no involvement of the nervous system. |
| N/L | N/L | N/L | N/L | N/L | Dysfibrinogenemia, congenital (DYSFIBRIN | A disorder characterized by qualitative abnormalities (dysfibrinogenemia) of the circulating fibrinogen. Affected individuals are frequently asymptomatic, but some patients have bleeding diathesis, thromboembolic complications, or both. In some cases, dysfibrinogenemia is associated with low circulating fibrinogen levels |
| Fibrinogen beta chain | FGB | P02675 | https://www.uniprot.org/uniprot/P02675 | HQLYIDETVNSNIPTNLR | Congenital afibrinogenemia (CAFBN) | Rare autosomal recessive disorder is characterized by bleeding that varies from mild to severe and by complete absence or extremely low levels of plasma and platelet fibrinogen. |
| N/L | N/L | N/L | N/L | N/L | Dysfibrinogenemia, congenital (DYSFIBRIN | A disorder characterized by qualitative abnormalities (dysfibrinogenemia) of the circulating fibrinogen. Affected individuals are frequently asymptomatic, but some patients have bleeding diathesis, thromboembolic complications, or both. In some cases, dysfibrinogenemia is associated with low circulating fibrinogen levels |
| Fibrinogen gamma chain | FGG PRO2061 | P02679 | https://www.uniprot.org/uniprot/P02679 | YEASILTHDSSIR | Congenital afibrinogenemia (CAFBN) | Rare autosomal recessive disorder is characterized by bleeding that varies from mild to severe and by complete absence or extremely low levels of plasma and platelet fibrinogen. |
| N/L | N/L | N/L | N/L | N/L | Dysfibrinogenemia, congenital (DYSFIBRIN | A disorder characterized by qualitative abnormalities (dysfibrinogenemia) of the circulating fibrinogen. Affected individuals are frequently asymptomatic, but some patients have bleeding diathesis, thromboembolic complications, or both. In some cases, dysfibrinogenemia is associated with low circulating fibrinogen levels |
| Fibronectin | FN1 FN | P02751 | https://www.uniprot.org/uniprot/P02751 | HTSVQTTSSGSGPFTDVR | Glomerulopathy with fibronectin deposits 2 (GFND2) | Genetically heterogeneous autosomal dominant disorder characterized clinically by proteinuria, microscopic hematuria, and hypertension that leads to end-stage renal failure in the second to fifth decade of life. |
| N/L | N/L | N/L | N/L | N/L | Spondylometaphyseal dysplasia, corner fracture type (SMDCF) | An autosomal dominant form of spondylometaphyseal dysplasia, a group of short stature disorders distinguished by abnormalities in the vertebrae and the metaphyses of the tubular bones. SMDCF is characterized by flake-like, triangular, or curvilinear ossification centers at the edges of irregular metaphyses that simulate fractures. These corner fractures involve the distal tibia, the ulnar aspect of the distal radius, the proximal humerus, and the proximal femur. They represent irregular ossification at the growth plates and secondary ossification centers |
| Fibulin-1 | FBLN1 PP213 | P23142 | https://www.uniprot.org/uniprot/P23142 | TGYYFDGISR | metacarpal and metatarsal synostoses | A chromosomal aberration involving FBLN1 is found in a complex type of synpolydactyly referred to as 3/3-prime/4 synpolydactyly associated with metacarpal and metatarsal synostoses. Reciprocal translocation t(12;22)(p11.2;q13.3) with RASSF8. Fibroblasts derived from a patient with synpolydactyly displayed alterations in the level of isoform D splice variant incorporated into the ECM and secreted into the conditioned culture medium. By contrast, the expression of isoform C was not perturbed in the patients fibroblasts. Furthermore, no aberrant polypeptides were detected in extracts of cultured patients fibroblasts. The translocation t(12;22) may result in haploinsufficiency of the isoform D splice variant, which could lead to the observed limb malformation. {ECO:0000269|PubMed:11836357}.; DISEASE: Note=Elevated expression and altered processing of FBLN1 protein is associated with human breast cancer. { |
| N/L | N/L | N/L | N/L | N/L | Human Breast Cancer | Note=Elevated expression and altered processing of FBLN1 protein is associated with human breast cancer. |
| Ficolin-2 | FCN2 FCNL | Q15485 | https://www.uniprot.org/uniprot/Q15485 | GTHGSFANGINWK | 0 | N/L |
| Ficolin-3 | FCN3 FCNH HAKA1 | O75636 | https://www.uniprot.org/uniprot/O75636 | YAVSEAAAHK | Ficolin 3 deficiency (FCN3D) | A disorder characterized by immunodeficiency, recurrent infections, brain abscesses and recurrent warts on the fingers. Affected individuals have normal levels of lymphocytes, normal T-cell responses, and normal antibodies, but a selective deficient antibody response to pneumococcal polysaccharide vaccine |
| Follistatin-related protein 1 | FSTL1 FRP | Q12841 | https://www.uniprot.org/uniprot/Q12841 | YVQELQK | 0 | N/L |
| Fructose-bisphosphate aldolase B | ALDOB ALDB | P05062 | https://www.uniprot.org/uniprot/P05062 | ALQASALAAWGGK | Hereditary fructose intolerance (HFI) | Autosomal recessive disease that results in an inability to metabolize fructose and related sugars. Complete exclusion of fructose results in dramatic recovery; however, if not treated properly, HFI subjects suffer episodes of hypoglycemia, general ill condition, and risk of death the remainder of life. |
| Galectin-3 | LGALS3 MAC2 | P17931 | https://www.uniprot.org/uniprot/P17931 | IALDFQR | 0 | N/L |
| Galectin-3-binding protein | LGALS3BP M2BP | Q08380 | https://www.uniprot.org/uniprot/Q08380 | SDLAVPSELALLK | 0 | N/L |
| Gamma-enolase | ENO2 | P09104 | https://www.uniprot.org/uniprot/P09104 | YITGDQLGALYQDFVR | 0 | N/L |
| Gelsolin | GSN | P06396 | https://www.uniprot.org/uniprot/P06396 | AGALNSNDAFVLK | Amyloidosis 5 (AMYL5) | A hereditary generalized amyloidosis due to gelsolin amyloid deposition. It is typically characterized by cranial neuropathy and lattice corneal dystrophy. Most patients have modest involvement of internal organs, but severe systemic disease can develop in some individuals causing peripheral polyneuropathy, amyloid cardiomyopathy, and nephrotic syndrome leading to renal failure.. |
| Glial fibrillary acidic protein | GFAP | P14136 | https://www.uniprot.org/uniprot/P14136 | LADVYQAELR | Alexander disease (ALXDRD) | A rare disorder of the central nervous system. The most common form affects infants and young children, and is characterized by progressive failure of central myelination, usually leading to death within the first decade. Infants with Alexander disease develop a leukodystrophy with macrocephaly, seizures, and psychomotor retardation. Patients with juvenile or adult forms typically experience ataxia, bulbar signs and spasticity, and a more slowly progressive course. Histologically, Alexander disease is characterized by Rosenthal fibers, homogeneous eosinophilic inclusions in astrocytes. |
| Glutamate receptor ionotropic NMDA 2A | GRIN2A NMDAR2A | Q12879 | https://www.uniprot.org/uniprot/Q12879 | FSYIPEAK | Epilepsy, focal, with speech disorder and with or without mental retardation (FESD) | A highly variable neurologic disorder with features ranging from severe early-onset seizures associated with delayed psychomotor development, persistent speech difficulties, and mental retardation to a more benign entity characterized by childhood onset of mild or asymptomatic seizures associated with transient speech difficulties followed by remission of seizures in adolescence and normal psychomotor development. The disorder encompasses several clinical entities, including Landau-Kleffner syndrome, epileptic encephalopathy with continuous spike and wave during slow-wave sleep, autosomal dominant rolandic epilepsy, mental retardation and speech dyspraxia, and benign epilepsy with centrotemporal spikes. |
| Glutamate receptor ionotropic NMDA 2B | GRIN2B NMDAR2B | Q13224 | https://www.uniprot.org/uniprot/Q13224 | EPGGPSFTIGK | Mental retardation, autosomal dominant 6, with or without seizures (MRD6) | A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD6 additional features may include seizures, hypotonia, abnormal movements, such as dystonia, and autistic features |
| N/L | N/L | N/L | N/L | N/L | Epileptic encephalopathy, early infantile, 27 (EIEE27) | A form of epileptic encephalopathy, a heterogeneous group of severe childhood onset epilepsies characterized by refractory seizures, neurodevelopmental impairment, and poor prognosis. Development is normal prior to seizure onset, after which cognitive and motor delays become apparent. |
| N/L | N/L | N/L | N/L | N/L | N/L | A chromosomal aberrations involving GRIN2B has been found in patients with mental retardation. Translocations t(9;12)(p23;p13.1) and t(10;12)(q21.1;p13.1) with a common breakpoint in 12p13.1. |
| Glutathione peroxidase 3 | GPX3 GPXP | P22352 | https://www.uniprot.org/uniprot/P22352 | QEPGENSEILPTLK | 0 | N/L |
| Glutathione S-transferase P | GSTP1 FAEES3 GST3 | P09211 | https://www.uniprot.org/uniprot/P09211 | TLGLYGK | 0 | N/L |
| Haptoglobin | HP | P00738 | https://www.uniprot.org/uniprot/P00738 | DIAPTLTLYVGK | Anhaptoglobinemia (AHP) | A condition characterized by the absence of the serum glycoprotein haptoglobin. Serum levels of haptoglobin vary among normal persons: levels are low in the neonatal period and in the elderly, differ by population, and can be influenced by environmental factors, such as infection. Secondary hypohaptoglobinemia can occur as a consequence of hemolysis, during which haptoglobin binds to free hemoglobin. Congenital haptoglobin deficiency is a risk factor for anaphylactic non-hemolytic transfusion reactions. |
| Heat shock protein beta-1 | HSPB1 HSP27 HSP28 | P04792 | https://www.uniprot.org/uniprot/P04792 | LFDQAFGLPR | Charcot-Marie-Tooth disease 2F (CMT2F) | A dominant axonal form of Charcot-Marie-Tooth disease, a disorder of the peripheral nervous system, characterized by progressive weakness and atrophy, initially of the peroneal muscles and later of the distal muscles of the arms. Charcot-Marie-Tooth disease is classified in two main groups on the basis of electrophysiologic properties and histopathology: primary peripheral demyelinating neuropathies (designated CMT1 when they are dominantly inherited) and primary peripheral axonal neuropathies (CMT2). Neuropathies of the CMT2 group are characterized by signs of axonal degeneration in the absence of obvious myelin alterations, normal or slightly reduced nerve conduction velocities, and progressive distal muscle weakness and atrophy. Onset of Charcot-Marie-Tooth disease type 2F is between 15 and 25 years with muscle weakness and atrophy usually beginning in feet and legs (peroneal distribution). Upper limb involvement occurs later. |
| N/L | N/L | N/L | N/L | N/L | Neuronopathy, distal hereditary motor, 2B (HMN2B) | A neuromuscular disorder. Distal hereditary motor neuronopathies constitute a heterogeneous group of neuromuscular disorders caused by selective degeneration of motor neurons in the anterior horn of the spinal cord, without sensory deficit in the posterior horn. The overall clinical picture consists of a classical distal muscular atrophy syndrome in the legs without clinical sensory loss. The disease starts with weakness and wasting of distal muscles of the anterior tibial and peroneal compartments of the legs. Later on, weakness and atrophy may expand to the proximal muscles of the lower limbs and/or to the distal upper limbs |
| Hemoglobin subunit alpha | HBA1; HBA2 | P69905 | https://www.uniprot.org/uniprot/P69905 | VGAHAGEYGAEALER | Heinz body anemias (HEIBAN) | Form of non-spherocytic hemolytic anemia of Dacie type 1. After splenectomy, which has little benefit, basophilic inclusions called Heinz bodies are demonstrable in the erythrocytes. Before splenectomy, diffuse or punctate basophilia may be evident. Most of these cases are probably instances of hemoglobinopathy. The hemoglobin demonstrates heat lability. Heinz bodies are observed also with the Ivemark syndrome (asplenia with cardiovascular anomalies) and with glutathione peroxidase deficiency. . Note=The disease is caused by mutations affecting the gene represented in this entry. |
| N/L | N/L | N/L | N/L | N/L | Alpha-thalassemia (A-THAL) | A form of thalassemia. Thalassemias are common monogenic diseases occurring mostly in Mediterranean and Southeast Asian populations. The hallmark of alpha-thalassemia is an imbalance in globin-chain production in the adult HbA molecule. The level of alpha chain production can range from none to very nearly normal levels. Deletion of both copies of each of the two alpha-globin genes causes alpha(0)-thalassemia, also known as homozygous alpha thalassemia. Due to the complete absence of alpha chains, the predominant fetal hemoglobin is a tetramer of gamma-chains (Bart hemoglobin) that has essentially no oxygen carrying capacity. This causes oxygen starvation in the fetal tissues leading to prenatal lethality or early neonatal death. The loss of two alpha genes results in mild alpha-thalassemia, also known as heterozygous alpha-thalassemia. Affected individuals have small red cells and a mild anemia (microcytosis). If three of the four alpha-globin genes are functional, individuals are completely asymptomatic. Some rare forms of alpha-thalassemia are due to point mutations (non-deletional alpha-thalassemia) |
| N/L | N/L | N/L | N/L | N/L | non-immune hydrops fetalis | Note=Alpha(0)-thalassemia is associated with non-immune hydrops fetalis, a generalized edema of the fetus with fluid accumulation in the body cavities due to non-immune causes. Non-immune hydrops fetalis is not a diagnosis in itself but a symptom, a feature of many genetic disorders, and the end-stage of a wide variety of disorders |
| N/L | N/L | N/L | N/L | N/L | Hemoglobin H disease (HBH) | A form of alpha-thalassemia due to the loss of three alpha genes. This results in high levels of a tetramer of four beta chains (hemoglobin H), causing a severe and life-threatening anemia. Untreated, most patients die in childhood or early adolescence. |
| Hemopexin | HPX | P02790 | https://www.uniprot.org/uniprot/P02790 | NFPSPVDAAFR | 0 | N/L |
| Heparin cofactor 2 | SERPIND1 HCF2 | P05546 | https://www.uniprot.org/uniprot/P05546 | TLEAQLTPR | Thrombophilia due to heparin cofactor 2 deficiency (THPH10) | A hemostatic disorder characterized by a tendency to recurrent thrombosis |
| Hepatocyte growth factor-like protein | MST1 D3F15S2 DNF15S2 HGFL | P26927 | https://www.uniprot.org/uniprot/P26927 | SPLNDFQVLR | inflammatory bowel disease (IBD) | Note=MST1 variant Cys-689 may be associated with inflammatory bowel disease (IBD), a chronic, relapsing inflammation of the gastrointestinal tract with a complex etiology. It is unsure whether Cys-689 itself or a variation in linkage disequilibrium with Cys-689 is responsible for the association with IBD. |
| Histidine-rich glycoprotein | HRG | P04196 | https://www.uniprot.org/uniprot/P04196 | ADLFYDVEALDLESPK | Thrombophilia due to histidine-rich glycoprotein deficiency (THPH11) | A hemostatic disorder characterized by a tendency to thrombosis |
| Hornerin | HRNR S100A18 | Q86YZ3 | https://www.uniprot.org/uniprot/Q86YZ3 | GSGSGQSPSSGQHGTGFGR | 0 | N/L |
| Hyaluronan-binding protein 2 | HABP2 HGFAL PHBP | Q14520 | https://www.uniprot.org/uniprot/Q14520 | VVLGDQDLK | Thyroid cancer, non-medullary, 5 (NMTC5) | A form of non-medullary thyroid cancer (NMTC), a cancer characterized by tumors originating from the thyroid follicular cells. NMTCs represent approximately 95% of all cases of thyroid cancer and are classified into papillary, follicular, Hurthle cell, and anaplastic neoplasms. |
| Ig gamma-1 chain C region | IGHG1 | P01857 | https://www.uniprot.org/uniprot/P01857 | GPSVFPLAPSSK | Multiple myeloma (MM) | A malignant tumor of plasma cells usually arising in the bone marrow and characterized by diffuse involvement of the skeletal system, hyperglobulinemia, Bence-Jones proteinuria and anemia. Complications of multiple myeloma are bone pain, hypercalcemia, renal failure and spinal cord compression. The aberrant antibodies that are produced lead to impaired humoral immunity and patients have a high prevalence of infection. Amyloidosis may develop in some patients. Multiple myeloma is part of a spectrum of diseases ranging from monoclonal gammopathy of unknown significance (MGUS) to plasma cell leukemia. |
| Ig mu chain C region | IGHM | P01871 | https://www.uniprot.org/uniprot/P01871 | GFPSVLR | NA | N/L |
| N/L | IGHM | P01871 | https://www.uniprot.org/uniprot/P01871 | VSVFVPPR | Agammaglobulinemia 1, autosomal recessive (AGM1) | A primary immunodeficiency characterized by profoundly low or absent serum antibodies and low or absent circulating B cells due to an early block of B-cell development. Affected individuals develop severe infections in the first years of life. |
| IgGFc-binding protein | FCGBP | Q9Y6R7 | https://www.uniprot.org/uniprot/Q9Y6R7 | GATTSPGVYELSSR | 0 | N/L |
| Immunoglobulin kappa variable 4-1 | IGKV4-1 | P06312 | https://www.uniprot.org/uniprot/P06312 | NYLAWYQQKPGQPPK | 0 | N/L |
| Insulin-like growth factor I | IGF1 IBP1 | P05019 | https://www.uniprot.org/uniprot/P05019 | GFYFNKPTGYGSSSR | Insulin-like growth factor I deficiency (IGF1 deficiency) | Autosomal recessive disorder characterized by growth retardation, sensorineural deafness and mental retardation. |
| Insulin-like growth factor-binding protein 1 | IGFBP1 IBP1 | P08833 | https://www.uniprot.org/uniprot/P08833 | ALPGEQQPLHALTR | 0 | N/L |
| Insulin-like growth factor-binding protein 2 | IGFBP2 BP2 IBP2 | P18065 | https://www.uniprot.org/uniprot/P18065 | LIQGAPTIR | 0 | N/L |
| Insulin-like growth factor-binding protein 3 | IGFBP3 IBP3 | P17936 | https://www.uniprot.org/uniprot/P17936 | FLNVLSPR | 0 | N/L |
| Insulin-like growth factor-binding protein complex acid labile subunit | IGFALS ALS | P35858 | https://www.uniprot.org/uniprot/P35858 | LAELPADALGPLQR | Acid-labile subunit deficiency (ACLSD) | A disorder characterized by severely reduced serum IGF-I and IGFBP-3 concentrations and mild growth retardation. Pubertal delay in boys and insulin insensitivity are common findings. |
| Inter-alpha-trypsin inhibitor heavy chain H1 | ITIH1 IGHEP1 | P19827 | https://www.uniprot.org/uniprot/P19827 | GSLVQASEANLQAAQDFVR | 0 | N/L |
| Inter-alpha-trypsin inhibitor heavy chain H2 | ITIH2 IGHEP2 | P19823 | https://www.uniprot.org/uniprot/P19823 | SLAPTAAAK | 0 | N/L |
| Inter-alpha-trypsin inhibitor heavy chain H4 | ITIH4 IHRP ITIHL1 PK120 PRO1851 | Q14624 | https://www.uniprot.org/uniprot/Q14624 | SPEQQETVLDGNLIIR | 0 | N/L |
| Intercellular adhesion molecule 1 | ICAM1 | P05362 | https://www.uniprot.org/uniprot/P05362 | LLGIETPLPK | 0 | N/L |
| Interleukin-10 | IL10 | P22301 | https://www.uniprot.org/uniprot/P22301 | AHVNSLGENLK | 0 | N/L |
| Interleukin-6 | IL6 IFNB2 | P05231 | https://www.uniprot.org/uniprot/P05231 | FESSEEQAR | Rheumatoid arthritis systemic juvenile (RASJ) | An inflammatory articular disorder with systemic-onset beginning before the age of 16. It represents a subgroup of juvenile arthritis associated with severe extraarticular features and occasionally fatal complications. During active phases of the disorder, patients display a typical daily spiking fever, an evanescent macular rash, lymphadenopathy, hepatosplenomegaly, serositis, myalgia and arthritiS |
| N/L | N/L | N/L | N/L | N/L | Kaposi sarcoma | A IL6 promoter polymorphism is associated with a lifetime risk of development of Kaposi sarcoma in HIV-infected men. |
| Interstitial collagenase | MMP1 CLG | P03956 | https://www.uniprot.org/uniprot/P03956 | AFQLWSNVTPLTFTK | 0 | N/L |
| Kallistatin | SERPINA4 KST PI4 | P29622 | https://www.uniprot.org/uniprot/P29622 | VGSALFLSHNLK | 0 | N/L |
| Keratin type I cytoskeletal 10 | KRT10 KPP | P13645 | https://www.uniprot.org/uniprot/P13645 | SLLEGEGSSGGGGR | Epidermolytic hyperkeratosis (EHK) | An autosomal dominant skin disorder characterized by widespread blistering and an ichthyotic erythroderma at birth that persist into adulthood. Histologically there is a diffuse epidermolytic degeneration in the lower spinous layer of the epidermis. Within a few weeks from birth, erythroderma and blister formation diminish and hyperkeratoses develop. |
| N/L | N/L | N/L | N/L | N/L | Ichthyosis annular epidermolytic (AEI) | A skin disorder resembling bullous congenital ichthyosiform erythroderma. Affected individuals present with bullous ichthyosis in early childhood and hyperkeratotic lichenified plaques in the flexural areas and extensor surfaces at later ages. The feature that distinguishes AEI from BCIE is dramatic episodes of flares of annular polycyclic plaques with scale, which coalesce to involve most of the body surface and can persist for several weeks or even months. |
| N/L | N/L | N/L | N/L | N/L | Erythroderma, ichthyosiform, congenital reticular (CRIE) | A rare skin condition characterized by slowly enlarging islands of normal skin surrounded by erythematous ichthyotic patches in a reticulated pattern. The condition starts in infancy as a lamellar ichthyosis, with small islands of normal skin resembling confetti appearing in late childhood and at puberty. Histopathologic findings include band-like parakeratosis, psoriasiform acanthosis, and vacuolization of keratinocytes with binucleated cells in the upper epidermis, sometimes associated with amyloid deposition in the dermis. Ultrastructural abnormalities include perinuclear shells formed from a network of fine filaments in the upper epidermis. |
| Keratin type I cytoskeletal 9 | KRT9 | P35527 | https://www.uniprot.org/uniprot/P35527 | TLLDIDNTR | Keratoderma, palmoplantar, epidermolytic (EPPK) | A dermatological disorder characterized by diffuse thickening of the epidermis on the entire surface of palms and soles sharply bordered with erythematous margins. Some patients may present knuckle pads, thick pads of skin appearing over the proximal phalangeal joints. |
| Keratin-type II cytoskeletal 2 epidermal | KRT2 KRT2A KRT2E | P35908 | https://www.uniprot.org/uniprot/P35908 | YEELQVTVGR | Ichthyosis bullosa of Siemens (IBS) | A rare autosomal dominant skin disorder displaying a type of epidermolytic hyperkeratosis characterized by generalized erythema and extensive blistering from birth. Large, dark gray hyperkeratoses are observed in later weeks. The skin of IBS patients is unusually fragile and has a tendency to shed the outer layers of the epidermis, producing localized denuded areas (molting effect). IBS usually improves with age so that in most middle-aged patients the hyperkeratosis and keratotic lichenification is limited to the flexural folds of the major joints. |
| Kininogen-1 | KNG1 BDK KNG | P01042 | https://www.uniprot.org/uniprot/P01042 | DIPTNSPELEETLTHTITK | High molecular weight kininogen deficiency (HMWK deficiency) | Autosomal recessive coagulation defect. Patients with HWMK deficiency do not have a hemorrhagic tendency, but they exhibit abnormal surface-mediated activation of fibrinolysis. Note=The disease is caused by mutations affecting the gene represented in this entry. |
| L-selectin | SELL LNHR LYAM1 | P14151 | https://www.uniprot.org/uniprot/P14151 | AEIEYLEK | 0 | N/L |
| Lactotransferrin | LTF GIG12 LF | P02788 | https://www.uniprot.org/uniprot/P02788 | YLGPQYVAGITNLK | 0 | N/L |
| Leucine-rich alpha-2-glycoprotein | LRG1 LRG | P02750 | https://www.uniprot.org/uniprot/P02750 | DLLLPQPDLR | 0 | N/L |
| Lipopolysaccharide-binding protein | LBP | P18428 | https://www.uniprot.org/uniprot/P18428 | ITLPDFTGDLR | 0 | N/L |
| Lumican | LUM LDC SLRR2D | P51884 | https://www.uniprot.org/uniprot/P51884 | SLEDLQLTHNK | 0 | N/L |
| Lysozyme C | LYZ LZM | P61626 | https://www.uniprot.org/uniprot/P61626 | AWVAWR | Amyloidosis 8 (AMYL8) | A form of hereditary generalized amyloidosis. Clinical features include extensive visceral amyloid deposits, renal amyloidosis resulting in nephrotic syndrome, arterial hypertension, hepatosplenomegaly, cholestasis, petechial skin rash. There is no involvement of the nervous system. |
| Mannan-binding lectin serine protease 1 | MASP1 CRARF CRARF1 PRSS5 | P48740 | https://www.uniprot.org/uniprot/P48740 | TGVITSPDFPNPYPK | 3MC syndrome 1 (3MC1) | A form of 3MC syndrome, an autosomal recessive disorder characterized by facial dysmorphism, craniosynostosis, learning disability, and genital, limb and vesicorenal anomalies. Facial features include hypertelorism, blepharophimosis, blepharoptosis and highly arched eyebrows, cleft lip and/or palate. The term 3MC syndrome includes Carnevale, Mingarelli, Malpuech, and Michels syndromes. |
| Mannan-binding lectin serine protease 2A | MASP2 | O00187 | https://www.uniprot.org/uniprot/O00187 | WPEPVFGR | MASP2 deficiency (MASPD) | A disorder that results in autoimmune manifestations, recurrent severe infections, and chronic inflammatory disease. |
| Mannose-binding protein C | MBL2 COLEC1 MBL | P11226 | https://www.uniprot.org/uniprot/P11226 | WLTFSLGK | 0 | N/L |
| Matrix Gla protein | MGP MGLAP GIG36 | P08493 | https://www.uniprot.org/uniprot/P08493 | NANTFISPQQR | Keutel syndrome (KTLS) | An autosomal recessive disorder characterized by abnormal cartilage calcification, peripheral pulmonary stenosis neural hearing loss and midfacial hypoplasia. |
| Matrix metalloproteinase-9 | MMP9 CLG4B | P14780 | https://www.uniprot.org/uniprot/P14780 | AVIDDAFAR | Intervertebral disc disease (IDD) | A common musculo-skeletal disorder caused by degeneration of intervertebral disks of the lumbar spine. It results in low-back pain and unilateral leg pain. |
| N/L | N/L | N/L | N/L | N/L | Metaphyseal anadysplasia 2 (MANDP2) | A bone development disorder characterized by skeletal anomalies that resolve spontaneously with age. Clinical characteristics are evident from the first months of life and include slight shortness of stature and a mild varus deformity of the legs. Patients attain a normal stature in adolescence and show improvement or complete resolution of varus deformity of the legs and rhizomelic micromelia. |
| Melanotransferrin | MELTF MAP97 MFI2 | P08582 | https://www.uniprot.org/uniprot/P08582 | YYDYSGAFR | 0 | N/L |
| Metalloproteinase inhibitor 1 | TIMP1 CLGI TIMP | P01033 | https://www.uniprot.org/uniprot/P01033 | GFQALGDAADIR | 0 | N/L |
| Metalloproteinase inhibitor 2 | TIMP2 | P16035 | https://www.uniprot.org/uniprot/P16035 | EYLIAGK | 0 | N/L |
| Metalloproteinase inhibitor 4 | TIMP4 | Q99727 | https://www.uniprot.org/uniprot/Q99727 | VVPASADPADTEK | 0 | N/L |
| Microtubule-associated protein tau | MAPT MAPTL MTBT1 TAU | P10636 | https://www.uniprot.org/uniprot/P10636 | EADLPEPSEK | N/L | Note=In Alzheimer disease, the neuronal cytoskeleton in the brain is progressively disrupted and replaced by tangles of paired helical filaments (PHF) and straight filaments, mainly composed of hyperphosphorylated forms of TAU (PHF-TAU or AD P-TAU). O-GlcNAcylation is greatly reduced in Alzheimer disease brain cerebral cortex leading to an increase in TAU/MAPT phosphorylations |
| N/L | N/L | N/L | N/L | N/L | Frontotemporal dementia (FTD) | A form of dementia characterized by pathologic finding of frontotemporal lobar degeneration, presenile dementia with behavioral changes, deterioration of cognitive capacities and loss of memory. In some cases, parkinsonian symptoms are prominent. Neuropathological changes include frontotemporal atrophy often associated with atrophy of the basal ganglia, substantia nigra, amygdala. In most cases, protein tau deposits are found in glial cells and/or neurons. |
| N/L | N/L | N/L | N/L | N/L | Pick disease of the brain (PIDB) | A rare form of dementia pathologically defined by severe atrophy, neuronal loss and gliosis. It is characterized by the occurrence of tau-positive inclusions, swollen neurons (Pick cells) and argentophilic neuronal inclusions known as Pick bodies that disproportionally affect the frontal and temporal cortical regions. Clinical features include aphasia, apraxia, confusion, anomia, memory loss and personality deterioration. |
| N/L | N/L | N/L | N/L | N/L | corticobasal degeneration (CBD) | Defects in MAPT are a cause of corticobasal degeneration (CBD). It is marked by extrapyramidal signs and apraxia and can be associated with memory loss. Neuropathologic features may overlap Alzheimer disease, progressive supranuclear palsy, and Parkinson disease.; |
| N/L | N/L | N/L | N/L | N/L | Progressive supranuclear palsy 1 (PSNP1) | Characterized by akinetic-rigid syndrome, supranuclear gaze palsy, pyramidal tract dysfunction, pseudobulbar signs and cognitive capacities deterioration. Neurofibrillary tangles and gliosis but no amyloid plaques are found in diseased brains. Most cases appear to be sporadic, with a significant association with a common haplotype including the MAPT gene and the flanking regions. Familial cases show an autosomal dominant pattern of transmission with incomplete penetrance; genetic analysis of a few cases showed the occurrence of tau mutations, including a deletion of Asn-613. |
| N/L | N/L | N/L | N/L | N/L | Parkinson-dementia syndrome (PARDE) | A syndrome characterized by parkinsonism, tremor, rigidity, dementia, ophthalmoparesis and pyramidal signs. Neurofibrillary degeneration occurs in the hippocampus, basal ganglia and brainstem nuclei. Note=The disease is caused by mutations affecting the gene represented in this entry. |
| Mucin-16 | MUC16 CA125 | Q8WXI7 | https://www.uniprot.org/uniprot/Q8WXI7 | ELGPYTLDR | 0 | N/L |
| Myelin basic protein | MBP | P02686 | https://www.uniprot.org/uniprot/P02686 | GVDAQGTLSK | chronical multiple sclerosis (MS) and Marburg disease | The reduction in the surface charge of citrullinated and/or methylated MBP could result in a weakened attachment to the myelin membrane. This mechanism could be operative in demyelinating diseases such as chronical multiple sclerosis (MS), and fulminating MS (Marburg disease). |
| Myeloblastin | PRTN3 MBN | P24158 | https://www.uniprot.org/uniprot/P24158 | LVNVVLGAHNVR | Wegener’s granulomatosis | Is the major autoantigen in anti-neutrophil cytoplasmic autoantibody (ANCA)-associated vasculitis (Wegener’s granulomatosis) (PubMed:2377228, PubMed:2679910). This complex, systemic disease is characterized by granulomatous inflammation with necrotizing lesions in the respiratory tract, glomerulonephritis, vasculitis, and anti-neutrophil cytoplasmatic autoantibodies detected in patient sera |
| Myeloperoxidase | MPO | P05164 | https://www.uniprot.org/uniprot/P05164 | VFFASWR | Myeloperoxidase deficiency (MPOD) | A disorder characterized by decreased myeloperoxidase activity in neutrophils and monocytes that results in disseminated candidiasis. |
| N-acetylmuramoyl-L-alanine amidase | PGLYRP2 PGLYRPL PGRPL UNQ3103/PRO10102 | Q96PD5 | https://www.uniprot.org/uniprot/Q96PD5 | AGLLRPDYALLGHR | 0 | N/L |
| N(G),N(G)-dimethylarginine dimethylaminohydrolase 1 | DDAH1 DDAH | O94760 | https://www.uniprot.org/uniprot/O94760 | TPEEYPESAK | 0 | N/L |
| Natriuretic peptides B | NPPB | P16860 | https://www.uniprot.org/uniprot/P16860 | EVATEGIR | 0 | N/L |
| Neuropilin-2 | NRP2 VEGF165R2 | O60462 | https://www.uniprot.org/uniprot/O60462 | ALQVVR | 0 | N/L |
| Neutrophil gelatinase-associated lipocalin | LCN2 HNL NGAL | P80188 | https://www.uniprot.org/uniprot/P80188 | ITLYGR | 0 | N/L |
| Nucleoside diphosphate kinase A | NME1 NDPKA NM23 | P15531 | https://www.uniprot.org/uniprot/P15531 | PFFAGLVK | 0 | N/L |
| Occludin | OCLN | Q16625 | https://www.uniprot.org/uniprot/Q16625 | SLQSELDEINK | Pseudo-TORCH syndrome 1 (PTORCH1) | An autosomal recessive neurologic disorder with characteristic clinical and neuroradiologic features that mimic intrauterine TORCH infection in the absence of evidence of infection. Affected individuals have congenital microcephaly, intracranial calcifications, and severe developmental delay. |
| Osteopontin | SPP1 BNSP OPN PSEC0156 | P10451 | https://www.uniprot.org/uniprot/P10451 | GDSVVYGLR | 0 | N/L |
| Oxidized low-density lipoprotein receptor 1 | OLR1 CLEC8A LOX1 | P78380 | https://www.uniprot.org/uniprot/P78380 | LEGQISAR | Alzheimer disease (AD) | Independent association genetic studies have implicated OLR1 gene variants in myocardial infarction susceptibility.; DISEASE: Note=OLR1 may be involved in Alzheimer disease (AD). Involvement in AD is however unclear: according to some authors |
| P-selectin | SELP GMRP GRMP | P16109 | https://www.uniprot.org/uniprot/P16109 | TWTWVGTK | Ischemic stroke (ISCHSTR) | A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. |
| Pappalysin-1 | PAPPA | Q13219 | https://www.uniprot.org/uniprot/Q13219 | AYLDVNELK | 0 | N/L |
| Peroxiredoxin-1 | PRDX1 PAGA PAGB TDPX2 | Q06830 | https://www.uniprot.org/uniprot/Q06830 | ADEGISFR | 0 | N/L |
| Peroxiredoxin-2 | PRDX2 NKEFB TDPX1 | P32119 | https://www.uniprot.org/uniprot/P32119 | GLFIIDGK | 0 | N/L |
| Phosphatidylcholine-sterol acyltransferase | LCAT | P04180 | https://www.uniprot.org/uniprot/P04180 | SSGLVSNAPGVQIR | Lecithin-cholesterol acyltransferase deficiency (LCATD) | A disorder of lipoprotein metabolism characterized by inadequate esterification of plasmatic cholesterol. Two clinical forms are recognized: complete LCAT deficiency and fish-eye disease. LCATD is generally referred to the complete form which is associated with absence of both alpha and beta LCAT activities resulting in esterification anomalies involving both HDL (alpha-LCAT activity) and LDL (beta-LCAT activity). It causes a typical triad of diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with renal failure.. |
| N/L | N/L | N/L | N/L | N/L | Fish-eye disease (FED) | A disorder of lipoprotein metabolism due to partial lecithin-cholesterol acyltransferase deficiency that affects only alpha-LCAT activity. FED is characterized by low plasma HDL and corneal opacities due to accumulation of cholesterol deposits in the cornea (‘fish-eye’). |
| Phosphatidylinositol-glycan-specific phospholipase D | GPLD1 PIGPLD1 | P80108 | https://www.uniprot.org/uniprot/P80108 | FGSSLITVR | 0 | N/L |
| Phospholipid transfer protein | PLTP | P55058 | https://www.uniprot.org/uniprot/P55058 | AVEPQLQEEER | 0 | N/L |
| Pigment epithelium-derived factor | SERPINF1 PEDF PIG35 | P36955 | https://www.uniprot.org/uniprot/P36955 | LQSLFDSPDFSK | Osteogenesis imperfecta 6 (OI6) | A form of osteogenesis imperfecta, a connective tissue disorder characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI6 is a severe, autosomal recessive form compatible with OI type III in the Sillence classification. |
| Plasma protease C1 inhibitor | SERPING1 C1IN C1NH | P05155 | https://www.uniprot.org/uniprot/P05155 | FQPTLLTLPR | Hereditary angioedema (HAE) | An autosomal dominant disorder characterized by episodic local swelling involving subcutaneous or submucous tissue of the upper respiratory and gastrointestinal tracts, face, extremities, and genitalia. Hereditary angioedema due to C1 esterase inhibitor deficiency is comprised of two clinically indistinguishable forms. In hereditary angioedema type 1, serum levels of C1 esterase inhibitor are decreased, while in type 2, the levels are normal or elevated, but the protein is non-functional. |
| Plasma serine protease inhibitor | SERPINA5 PCI PLANH3 PROCI | P05154 | https://www.uniprot.org/uniprot/P05154 | GFQQLLQELNQPR | 0 | N/L |
| Plasminogen | PLG | P00747 | https://www.uniprot.org/uniprot/P00747 | EAQLPVIENK | Plasminogen deficiency (PLGD) | A disorder characterized by decreased serum plasminogen activity. Two forms of the disorder are distinguished: type 1 deficiency is additionally characterized by decreased plasminogen antigen levels and clinical symptoms, whereas type 2 deficiency, also known as dysplasminogenemia, is characterized by normal, or slightly reduced antigen levels, and absence of clinical manifestations. Plasminogen deficiency type 1 results in markedly impaired extracellular fibrinolysis and chronic mucosal pseudomembranous lesions due to subepithelial fibrin deposition and inflammation. The most common clinical manifestation of type 1 deficiency is ligneous conjunctivitis in which pseudomembranes formation on the palpebral surfaces of the eye progresses to white, yellow-white, or red thick masses with a wood-like consistency that replace the normal mucosa. |
| Plasminogen activator inhibitor 1 | SERPINE1 PAI1 PLANH1 | P05121 | https://www.uniprot.org/uniprot/P05121 | VFQQVAQASK | Plasminogen activator inhibitor-1 deficiency (PAI-1D) | A hematologic disorder characterized by increased bleeding after trauma, injury, or surgery. Affected females have menorrhagia. The bleeding defect is due to increased fibrinolysis of fibrin blood clots due to deficiency of plasminogen activator inhibitor-1, which inhibits tissue and urinary activators of plasminogen. Note=The disease is caused by mutations affecting the gene represented in this entry. |
| Plastin-2 | LCP1 PLS2 | P13796 | https://www.uniprot.org/uniprot/P13796 | ISFDEFIK | B-cell non-Hodgkin lymphomas (B-cell NHL) | Chromosomal aberrations involving LCP1 is a cause of B-cell non-Hodgkin lymphomas (B-cell NHL)..; DISEASE: |
| N/L | N/L | N/L | N/L | N/L | isolated coloboma | Defects in LCP1 has been found in a patient with isolated coloboma, a defect of the eye characterized by the absence of ocular structures due to abnormal morphogenesis of the optic cup and stalk, and the fusion of the fetal fissure (optic fissure). Isolated colobomas may be associated with an abnormally small eye (microphthalmia) or small cornea. |
| Platelet endothelial cell adhesion molecule | PECAM1 | P16284 | https://www.uniprot.org/uniprot/P16284 | SELVTVTESFSTPK | 0 | N/L |
| Platelet glycoprotein VI | GP6 | Q9HCN6 | https://www.uniprot.org/uniprot/Q9HCN6 | EGDPAPYK | Bleeding disorder, platelet-type 11 (BDPLT11) | A mild to moderate bleeding disorder caused by defective platelet activation and aggregation in response to collagen. |
| Platelet-activating factor acetylhydrolase | PLA2G7 PAFAH | Q13093 | https://www.uniprot.org/uniprot/Q13093 | GSVHQNFADFTFATGK | Platelet-activating factor acetylhydrolase deficiency (PAFAD) | An enzymatic deficiency that results in exacerbated bodily response to inflammatory agents. It can be associated with several disease states including inflammatory gastrointestinal disorders, asthma and atopy. Asthmatic individuals with PAFAD may manifest aggravated respiratory symptoms |
| N/L | N/L | N/L | N/L | N/L | Asthma (ASTHMA) | The most common chronic disease affecting children and young adults. It is a complex genetic disorder with a heterogeneous phenotype, largely attributed to the interactions among many genes and between these genes and the environment. It is characterized by recurrent attacks of paroxysmal dyspnea, with wheezing due to spasmodic contraction of the bronchi. |
| N/L | N/L | N/L | N/L | N/L | Atopic hypersensitivity (ATOPY) | A condition characterized by predisposition to develop hypersensitivity reactions. Atopic individuals can develop eczema, allergic rhinitis and allergic asthma. |
| Pregnancy zone protein | PZP CPAMD6 | P20742 | https://www.uniprot.org/uniprot/P20742 | ISEITNIVSK | 0 | N/L |
| Proenkephalin-A | PENK | P01210 | https://www.uniprot.org/uniprot/P01210 | ELLETGDNR | 0 | N/L |
| Prolactin | PRL | P01236 | https://www.uniprot.org/uniprot/P01236 | IDNYLK | 0 | N/L |
| Protein AMBP | AMBP HCP ITIL | P02760 | https://www.uniprot.org/uniprot/P02760 | HHGPTITAK | 0 | N/L |
| Protein S100-A12 | S100A12 | P80511 | https://www.uniprot.org/uniprot/P80511 | GHFDTLSK | 0 | N/L |
| Protein S100-A9 | S100A9 CAGB CFAG MRP14 | P06702 | https://www.uniprot.org/uniprot/P06702 | DLQNFLK | 0 | N/L |
| Protein S100-B | S100B | P04271 | https://www.uniprot.org/uniprot/P04271 | EQEVVDK | 0 | N/L |
| Protein Z-dependent protease inhibitor | SERPINA10 ZPI UNQ707/PRO1358 | Q9UK55 | https://www.uniprot.org/uniprot/Q9UK55 | ETSNFGFSLLR | 0 | N/L |
| Protein_deglycase_DJ-1 | PARK7 | Q99497 | https://www.uniprot.org/uniprot/Q99497 | ALVILAK | Parkinson disease 7 (PARK7) | A neurodegenerative disorder characterized by resting tremor, postural tremor, bradykinesia, muscular rigidity, anxiety and psychotic episodes. PARK7 has onset before 40 years, slow progression and initial good response to levodopa. Some patients may show traits reminiscent of amyotrophic lateral sclerosis-parkinsonism/dementia complex (Guam disease). |
| Proteoglycan 4 | PRG4 MSF SZP | Q92954 | https://www.uniprot.org/uniprot/Q92954 | DQYYNIDVPSR | Camptodactyly-arthropathy-coxa vara-pericarditis syndrome (CACP) | : An autosomal recessive disorder characterized by the association of congenital or early-onset camptodactyly and non-inflammatory arthropathy with synovial hyperplasia. Individuals with CACP have normal appearing joints at birth but with advancing age develop joint failure, non-inflammatory synoviocyte hyperplasia and subintimal fibrosis of the synovial capsule. Some patients also manifest progressive coxa vara deformity and/or non-inflammatory pericardial or pleural effusions. |
| Prothrombin | F2 | P00734 | https://www.uniprot.org/uniprot/P00734 | ELLESYIDGR | Factor II deficiency (FA2D) | A very rare blood coagulation disorder characterized by mucocutaneous bleeding symptoms. The severity of the bleeding manifestations correlates with blood factor II levels. |
| N/L | N/L | N/L | N/L | N/L | Ischemic stroke (ISCHSTR) | A stroke is an acute neurologic event leading to death of neural tissue of the brain and resulting in loss of motor, sensory and/or cognitive function. Ischemic strokes, resulting from vascular occlusion, is considered to be a highly complex disease consisting of a group of heterogeneous disorders with multiple genetic and environmental risk factors. |
| N/L | N/L | N/L | N/L | N/L | Thrombophilia due to thrombin defect (THPH1) | A multifactorial disorder of hemostasis characterized by abnormal platelet aggregation in response to various agents and recurrent thrombi formation. common genetic variation in the 3-prime untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increased risk of venous thrombosis.; |
| N/L | N/L | N/L | N/L | N/L | Pregnancy loss, recurrent, 2 (RPRGL2) | A common complication of pregnancy, resulting in spontaneous abortion before the fetus has reached viability. The term includes all miscarriages from the time of conception until 24 weeks of gestation. Recurrent pregnancy loss is defined as 3 or more consecutive spontaneous abortions. |
| Ras GTPase-activating protein nGAP | RASAL2 NGAP | Q9UJF2 | https://www.uniprot.org/uniprot/Q9UJF2 | ETQSTPQSAPQVR | 0 | N/L |
| Resistin | RETN FIZZ3 HXCP1 RSTN UNQ407/PRO1199 | Q9HD89 | https://www.uniprot.org/uniprot/Q9HD89 | IQEVAGSLIFR | 0 | N/L |
| Retinol-binding protein 4 | RBP4 PRO2222 | P02753 | https://www.uniprot.org/uniprot/P02753 | YWGVASFLQK | Retinal dystrophy, iris coloboma, and comedogenic acne syndrome (RDCCAS) | A disease characterized by retinal degeneration, ocular colobomas involving both the anterior and posterior segment, impaired night vision and loss of visual acuity. Additional characteristic features include developmental abnormalities and severe acne. Loss of functional RBP4 protein results in serum retinol deficiency. Lack of normal levels of retinol impairs the visual cycle leading to night blindness at early stages; prolonged deficiency may lead to retinal degeneration. Additionally, retinol deficiency may result in dry skin, increased susceptibility to infection and acne |
| N/L | N/L | N/L | N/L | N/L | Microphthalmia, isolated, with coloboma, 10 (MCOPCB10) | A disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues. Ocular abnormalities like opacities of the cornea and lens, scaring of the retina and choroid, and other abnormalities may also be present. Ocular colobomas are a set of malformations resulting from abnormal morphogenesis of the optic cup and stalk, and the fusion of the fetal fissure (optic fissure). |
| Serotransferrin | TF PRO1400 | P02787 | https://www.uniprot.org/uniprot/P02787 | DGAGDVAFVK | Atransferrinemia (ATRAF) | A rare autosomal recessive disorder characterized by abnormal synthesis of transferrin leading to iron overload and microcytic hypochromic anemia. |
| Serum albumin | ALB GIG20 GIG42 PRO0903 PRO1708 PRO2044 PRO2619 PRO2675 UNQ696/PRO1341 | P02768 | https://www.uniprot.org/uniprot/P02768 | LVNEVTEFAK | Hyperthyroxinemia, familial dysalbuminemic (FDAH) | A disorder characterized by abnormally elevated levels of total serum thyroxine (T4) in euthyroid patients. It is due to abnormal serum albumin that binds T4 with enhanced affinity. |
| N/L | N/L | N/L | N/L | N/L | Analbuminemia (ANALBA) | A rare autosomal recessive disorder manifested by the presence of a very low amount of circulating serum albumin. Affected individuals manifest mild edema, hypotension, fatigue, and, occasionally, lower body lipodystrophy (mainly in adult females). The most common biochemical finding is hyperlipidemia, with a significant increase in the total and LDL cholesterol concentrations, but normal concentrations of HDL cholesterol and triglycerides. |
| Serum amyloid A-1 and A-2 proteins | SAA1 | P0DJI8 | https://www.uniprot.org/uniprot/P0DJI8 | EANYIGSDK | N/L | Note=Reactive, secondary amyloidosis is characterized by the extracellular accumulation in various tissues of the SAA1 protein. These deposits are highly insoluble and resistant to proteolysis; they disrupt tissue structure and compromise function. |
| N/L | N/L | N/L | N/L | N/L | N/L | Note=Elevated serum SAA1 protein levels may be associated with lung cancer. |
| Serum amyloid A-1 and A-2 proteins | SAA2 | P0DJI9 | https://www.uniprot.org/uniprot/P0DJI9 | EANYIGSDK | N/L | Note=Reactive, secondary amyloidosis is characterized by the extracellular accumulation in various tissues of the SAA2 protein. These deposits are highly insoluble and resistant to proteolysis; they disrupt tissue structure and compromise function. |
| Serum amyloid A-4 protein | SAA4 CSAA | P35542 | https://www.uniprot.org/uniprot/P35542 | GNYDAAQR | 0 | N/L |
| Serum amyloid P-component | APCS PTX2 | P02743 | https://www.uniprot.org/uniprot/P02743 | IVLGQEQDSYGGK | N/L | Note=SAP is a precursor of amyloid component P which is found in basement membrane and associated with amyloid deposits. |
| Serum paraoxonase/arylesterase 1 | PON1 PON | P27169 | https://www.uniprot.org/uniprot/P27169 | IFFYDSENPPASEVLR | Microvascular complications of diabetes 5 (MVCD5) | Pathological conditions that develop in numerous tissues and organs as a consequence of diabetes mellitus. They include diabetic retinopathy, diabetic nephropathy leading to end-stage renal disease, and diabetic neuropathy. Diabetic retinopathy remains the major cause of new-onset blindness among diabetic adults. It is characterized by vascular permeability and increased tissue ischemia and angiogenesis. Homozygosity for the Leu-55 allele is strongly associated with the development of retinal disease in diabetic patients. |
| Serum paraoxonase/lactonase 3 | PON3 | Q15166 | https://www.uniprot.org/uniprot/Q15166 | ILIGTVFHK | 0 | N/L |
| Sex hormone-binding globulin | SHBG | P04278 | https://www.uniprot.org/uniprot/P04278 | TSSSFEVR | 0 | N/L |
| SPARC | SPARC ON | P09486 | https://www.uniprot.org/uniprot/P09486 | LEAGDHPVELLAR | Osteogenesis imperfecta 17 (OI17) | An autosomal recessive form of osteogenesis imperfecta, a connective tissue disorder characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. |
| Spermine oxidase | SMOX C20orf16 SMO UNQ3039/PRO9854 | Q9NWM0 | https://www.uniprot.org/uniprot/Q9NWM0 | YYSTTHGALLSGQR | 0 | N/L |
| Sterile alpha motif domain-containing protein 9-like | SAMD9L C7orf6 DRIF2 KIAA2005 UEF | Q8IVG5 | https://www.uniprot.org/uniprot/Q8IVG5 | ENVLDEVANAK | Ataxia-pancytopenia syndrome (ATXPC) | An autosomal dominant disorder characterized by cerebellar ataxia, variable hematologic cytopenias, and predisposition to bone marrow failure and myeloid leukemia. |
| Stromelysin-1 | MMP3 STMY1 | P08254 | https://www.uniprot.org/uniprot/P08254 | TYFFVEDK | Coronary heart disease 6 (CHDS6) | A multifactorial disease characterized by an imbalance between myocardial functional requirements and the capacity of the coronary vessels to supply sufficient blood flow. Decreased capacity of the coronary vessels is often associated with thickening and loss of elasticity of the coronary arteries. A polymorphism in the MMP3 promoter region is associated with the risk of coronary heart disease and myocardial infarction, due to lower MMP3 proteolytic activity and higher extracellular matrix deposition in atherosclerotic lesions. |
| Target of Nesh-SH3 | ABI3BP NESHBP TARSH | Q7Z7G0 | https://www.uniprot.org/uniprot/Q7Z7G0 | IYLSDSLTGK | isolated coloboma | Note=Defects in ABI3BP has been found in a patient with isolated coloboma, a defect of the eye characterized by the absence of ocular structures due to abnormal morphogenesis of the optic cup and stalk, and the fusion of the fetal fissure (optic fissure). Isolated colobomas may be associated with an abnormally small eye (microphthalmia) or small cornea. |
| TBC1 domain family member 10A | TBC1D10A EPI64 TBC1D10 | Q9BXI6 | https://www.uniprot.org/uniprot/Q9BXI6 | YLPGYYSEK | 0 | N/L |
| Tenascin | TNC HXB | P24821 | https://www.uniprot.org/uniprot/P24821 | FTTDLDSPR | Deafness, autosomal dominant, 56 (DFNA56) | A form of non-syndromic sensorineural hearing loss. Sensorineural deafness results from damage to the neural receptors of the inner ear, the nerve pathways to the brain, or the area of the brain that receives sound information. DFNA56 is characterized by progressive hearing impairment with post-lingual onset. |
| Tenascin-X | TNXB HXBL TNX TNXB1 TNXB2 XB | P22105 | https://www.uniprot.org/uniprot/P22105 | ILISGLEPSTPYR | Ehlers-Danlos syndrome, classic-like (EDSCLL) | A form of Ehlers-Danlos syndrome, a group of connective tissue disorders characterized by skin hyperextensibility, articular hypermobility, and tissue fragility. EDSCLL patients lack atrophic scars, a major diagnostic criteria for classic Ehlers-Danlos syndrome. Delayed wound healing is only present in a subset of patients. EDSCLL inheritance is autosomal recessive. |
| N/L | N/L | N/L | N/L | N/L | Vesicoureteral reflux 8 (VUR8) | A disease belonging to the group of congenital anomalies of the kidney and urinary tract. It is characterized by the reflux of urine from the bladder into the ureters and sometimes into the kidneys, and is a risk factor for urinary tract infections. Primary disease results from a developmental defect of the ureterovesical junction. In combination with intrarenal reflux, the resulting inflammatory reaction may result in renal injury or scarring, also called reflux nephropathy. Extensive renal scarring impairs renal function and may predispose patients to hypertension, proteinuria, renal insufficiency and end-stage renal disease. |
| Putative tenascin-XA | TNXA XA | Q16473 | https://www.uniprot.org/uniprot/Q16473 | ILISGLEPSTPYR | 0 | N/L |
| Tetranectin | CLEC3B TNA | P05452 | https://www.uniprot.org/uniprot/P05452 | NWETEITAQPDGGK | 0 | N/L |
| Thrombomodulin | THBD THRM | P07204 | https://www.uniprot.org/uniprot/P07204 | SSVAADVISLLLNGDGGVGR | Thrombophilia due to thrombomodulin defect (THPH12) | A hemostatic disorder characterized by a tendency to thrombosis. The role of thrombomodulin in thrombosis is controversial. It is likely that genetic or environmental risk factors in addition to THBD variation are involved in the pathogenesis of venous thrombosis. |
| N/L | N/L | N/L | N/L | N/L | Hemolytic uremic syndrome atypical 6 (AHUS6) | An atypical form of hemolytic uremic syndrome. It is a complex genetic disease characterized by microangiopathic hemolytic anemia, thrombocytopenia, renal failure and absence of episodes of enterocolitis and diarrhea. In contrast to typical hemolytic uremic syndrome, atypical forms have a poorer prognosis, with higher death rates and frequent progression to end-stage renal disease. |
| Thrombospondin-1 | THBS1 TSP TSP1 | P07996 | https://www.uniprot.org/uniprot/P07996 | GTLLALER | 0 | N/L |
| Thrombospondin-4 | THBS4 TSP4 | P35443 | https://www.uniprot.org/uniprot/P35443 | KPQDFLEELK | 0 | N/L |
| Thyroglobulin | TG | P01266 | https://www.uniprot.org/uniprot/P01266 | FSPDDSAGASALLR | Thyroid dyshormonogenesis 3 (TDH3) | A disorder due to thyroid dyshormonogenesis, causing large goiters of elastic and soft consistency in the majority of patients. Although the degree of thyroid dysfunction varies considerably among patients with defective thyroglobulin synthesis, patients usually have a relatively high serum free triiodothyronine (T3) concentration with disproportionately low free tetraiodothyronine (T4) level. The maintenance of relatively high free T3 levels prevents profound tissue hypothyroidism except in brain and pituitary, which are dependent on T4 supply, resulting in neurologic and intellectual defects in some cases. |
| N/L | N/L | N/L | N/L | N/L | Autoimmune thyroid disease 3 (AITD3) | A complex autoimmune disorder comprising two related diseases affecting the thyroid: Graves disease and Hashimoto thyroiditis. In both disorders, thyroid-reactive T-cells are formed and infiltrate the thyroid gland. In Graves disease, the majority of the T-cells undergo a Th2 differentiation and activate B-cells to produce antibodies against the TSH receptor, which stimulate the thyroid and cause clinical hyperthyroidism. In contrast, Hashimoto thyroiditis is characterized by Th1 switching of the thyroid-infiltrating T-cells, which induces apoptosis of thyroid follicular cells and clinical hypothyroidism. |
| Thyroxine-binding globulin | SERPINA7 TBG | P05543 | https://www.uniprot.org/uniprot/P05543 | AVLHIGEK | 0 | N/L |
| Tissue_factor_pathway_inhibitor | TFPI LACI TFPI1 | P10646 | https://www.uniprot.org/uniprot/P10646 | FYYNSVIGK | 0 | N/L |
| Tissue-type plasminogen activator | PLAT | P00750 | https://www.uniprot.org/uniprot/P00750 | VVPGEEEQK | N/L | Increased activity of TPA results in increased fibrinolysis of fibrin blood clots that is associated with excessive bleeding. Defective release of TPA results in hypofibrinolysis that can lead to thrombosis or embolism. |
| Transcription factor SOX-11 | SOX11 | P35716 | https://www.uniprot.org/uniprot/P35716 | AAQSGDYGGAGDDYVLGSLR | Mental retardation, autosomal dominant 27 (MRD27) | A disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. MRD27 patients show dysmorphic facial features, microcephaly, growth deficiency, hypoplastic fifth toenails, and mild intellectual disability. |
| Transferrin receptor protein 1 | TFRC | P02786 | https://www.uniprot.org/uniprot/P02786 | GFVEPDHYVVVGAQR | Immunodeficiency 46 (IMD46) | An autosomal recessive primary immunodeficiency disorder characterized by early-onset chronic diarrhea, recurrent infections, hypo- or agammaglobulinemia, normal lymphocyte counts, intermittent neutropenia, and intermittent thrombocytopenia. |
| Transthyretin | TTR PALB | P02766 | https://www.uniprot.org/uniprot/P02766 | GSPAINVAVHVFR | Amyloidosis, transthyretin-related (AMYL-TTR) | A hereditary generalized amyloidosis due to transthyretin amyloid deposition. Protein fibrils can form in different tissues leading to amyloid polyneuropathies, amyloidotic cardiomyopathy, carpal tunnel syndrome, systemic senile amyloidosis. The disease includes leptomeningeal amyloidosis that is characterized by primary involvement of the central nervous system. Neuropathologic examination shows amyloid in the walls of leptomeningeal vessels, in pia arachnoid, and subpial deposits. Some patients also develop vitreous amyloid deposition that leads to visual impairment (oculoleptomeningeal amyloidosis). Clinical features include seizures, stroke-like episodes, dementia, psychomotor deterioration, variable amyloid deposition in the vitreous humor. |
| N/L | N/L | N/L | N/L | N/L | Hyperthyroxinemia, dystransthyretinemic (DTTRH) | A condition characterized by elevation of total and free thyroxine in healthy, euthyroid persons without detectable binding protein abnormalities. |
| N/L | N/L | N/L | N/L | N/L | Carpal tunnel syndrome 1 (CTS1) | A condition characterized by entrapment of the median nerve within the carpal tunnel. Symptoms include burning pain and paresthesias involving the ventral surface of the hand and fingers which may radiate proximally. Impairment of sensation in the distribution of the median nerve and thenar muscle atrophy may occur. This condition may be associated with repetitive occupational trauma, wrist injuries, amyloid neuropathies, rheumatoid arthritis. |
| Tumor necrosis factor receptor superfamily member 1A | TNFRSF1A TNFAR TNFR1 | P19438 | https://www.uniprot.org/uniprot/P19438 | LGLSDHEIDR | Familial hibernian fever (FHF) | A hereditary periodic fever syndrome characterized by recurrent fever, abdominal pain, localized tender skin lesions and myalgia. Reactive amyloidosis is the main complication and occurs in 25% of cases |
| N/L | N/L | N/L | N/L | N/L | Multiple sclerosis 5 (MS5) | A multifactorial, inflammatory, demyelinating disease of the central nervous system. Sclerotic lesions are characterized by perivascular infiltration of monocytes and lymphocytes and appear as indurated areas in pathologic specimens (sclerosis in plaques). The pathological mechanism is regarded as an autoimmune attack of the myelin sheath, mediated by both cellular and humoral immunity. Clinical manifestations include visual loss, extra-ocular movement disorders, paresthesias, loss of sensation, weakness, dysarthria, spasticity, ataxia and bladder dysfunction. Genetic and environmental factors influence susceptibility to the disease. Note=Disease susceptibility is associated with variations affecting the gene represented in this entry. An intronic mutation affecting alternative splicing and skipping of exon 6 directs increased expression of isoform 4 a transcript encoding a C-terminally truncated protein which is secreted and may function as a TNF antagonist. |
| Tumor necrosis factor receptor superfamily member 1B | TNFRSF1B TNFBR TNFR2 | P20333 | https://www.uniprot.org/uniprot/P20333 | DEQVPFSK | 0 | N/L |
| Vascular cell adhesion protein 1 | VCAM1 | P19320 | https://www.uniprot.org/uniprot/P19320 | NTVISVNPSTK | 0 | N/L |
| Vascular endothelial growth factor B | VEGFB VRF | P49765 | https://www.uniprot.org/uniprot/P49765 | VVSWIDVYTR | 0 | N/L |
| Vascular endothelial growth factor D | VEGFD FIGF | O43915 | https://www.uniprot.org/uniprot/O43915 | DLIQHPK | 0 | N/L |
| Vascular non-inflammatory molecule 3 | VNN3 | Q9NY84 | https://www.uniprot.org/uniprot/Q9NY84 | TETPVSK | 0 | N/L |
| Vasorin | VASN SLITL2 UNQ314/PRO357/PRO1282 | Q6EMK4 | https://www.uniprot.org/uniprot/Q6EMK4 | YLQGSSVQLR | 0 | N/L |
| Vitamin D-binding protein | GC | P02774 | https://www.uniprot.org/uniprot/P02774 | VLEPTLK | 0 | N/L |
| Vitamin K-dependent protein C | PROC | P04070 | https://www.uniprot.org/uniprot/P04070 | LGEYDLR | Thrombophilia due to protein C deficiency, autosomal dominant (THPH3) | A hemostatic disorder characterized by impaired regulation of blood coagulation and a tendency to recurrent venous thrombosis. Individuals with decreased amounts of protein C are classically referred to as having type I protein C deficiency and those with normal amounts of a functionally defective protein as having type II deficiency. |
| N/L | N/L | N/L | N/L | N/L | Thrombophilia due to protein C deficiency, autosomal recessive (THPH4) | A hemostatic disorder characterized by impaired regulation of blood coagulation and a tendency to recurrent venous thrombosis. It results in a thrombotic condition that can manifest as a severe neonatal disorder or as a milder disorder with late-onset thrombophilia. The severe form leads to neonatal death through massive neonatal venous thrombosis. Often associated with ecchymotic skin lesions which can turn necrotic called purpura fulminans, this disorder is very rare. |
| Vitamin K-dependent protein S | PROS1 PROS | P07225 | https://www.uniprot.org/uniprot/P07225 | VYFAGFPR | Thrombophilia due to protein S deficiency, autosomal dominant (THPH5) | A hemostatic disorder characterized by impaired regulation of blood coagulation and a tendency to recurrent venous thrombosis. Based on the plasma levels of total and free PROS1 as well as the serine protease-activated protein C cofactor activity, three types of THPH5 have been described: type I, characterized by reduced total and free PROS1 levels together with reduced anticoagulant activity; type III, in which only free PROS1 antigen and PROS1 activity levels are reduced; and the rare type II which is characterized by normal concentrations of both total and free PROS1 antigen, but low cofactor activity. |
| N/L | N/L | N/L | N/L | N/L | Thrombophilia due to protein S deficiency, autosomal recessive (THPH6) | A very rare and severe hematologic disorder resulting in thrombosis and secondary hemorrhage usually beginning in early infancy. Some affected individuals develop neonatal purpura fulminans, multifocal thrombosis, or intracranial hemorrhage. |
| Vitamin K-dependent protein Z | PROZ | P22891 | https://www.uniprot.org/uniprot/P22891 | DFAEHLLIPR | 0 | N/L |
| Vitronectin | VTN | P04004 | https://www.uniprot.org/uniprot/P04004 | FEDGVLDPDYPR | 0 | N/L |
| von Willebrand factor | VWF F8VWF | P04275 | https://www.uniprot.org/uniprot/P04275 | ILAGPAGDSNVVK | von Willebrand disease 1 (VWD1) | A common hemorrhagic disorder due to defects in von Willebrand factor protein and resulting in impaired platelet aggregation. Von Willebrand disease type 1 is characterized by partial quantitative deficiency of circulating von Willebrand factor, that is otherwise structurally and functionally normal. Clinical manifestations are mucocutaneous bleeding, such as epistaxis and menorrhagia, and prolonged bleeding after surgery or trauma. |
| N/L | N/L | N/L | N/L | N/L | von Willebrand disease 2 (VWD2) | A hemorrhagic disorder due to defects in von Willebrand factor protein and resulting in altered platelet aggregation. Von Willebrand disease type 2 is characterized by qualitative deficiency and functional anomalies of von Willebrand factor. It is divided in different subtypes including 2A, 2B, 2M and 2N (Normandy variant). The mutant VWF protein in types 2A, 2B and 2M are defective in their platelet-dependent function, whereas the mutant protein in type 2N is defective in its ability to bind factor VIII. Clinical manifestations are mucocutaneous bleeding, such as epistaxis and menorrhagia, and prolonged bleeding after surgery or trauma. |
| N/L | N/L | N/L | N/L | N/L | von Willebrand disease 3 (VWD3) | A severe hemorrhagic disorder due to a total or near total absence of von Willebrand factor in the plasma and cellular compartments, also leading to a profound deficiency of plasmatic factor VIII. Bleeding usually starts in infancy and can include epistaxis, recurrent mucocutaneous bleeding, excessive bleeding after minor trauma, and hemarthroses |
| Xaa-Pro dipeptidase | PEPD PRD | P12955 | https://www.uniprot.org/uniprot/P12955 | AVYEAVLR | Prolidase deficiency (PD) | A multisystem disorder associated with massive iminodipeptiduria and lack of or reduced prolidase activity in erythrocytes, leukocytes, or cultured fibroblasts. Clinical features include skin ulcers, developmental delay, recurrent infections, and a characteristic facies. |
| Zinc-alpha-2-glycoprotein | AZGP1 ZAG ZNGP1 | P25311 | https://www.uniprot.org/uniprot/P25311 | EIPAWVPFDPAAQITK | 0 | N/L |
| Protein Name | UniProt Accession Number |
|---|---|
| 78 kDa glucose-regulated protein | P11021 |
| Adipocyte plasma membrane-associated protein | Q9HDC9 |
| Adiponectin | Q15848 |
| Afamin | P43652 |
| Alpha-1-acid glycoprotein 1 | P02763 |
| Alpha-1-antichymotrypsin | P01011 |
| Alpha-1-antitrypsin | P01009 |
| Alpha-1B-glycoprotein_VAR_018369 | P04217 |
| Alpha-2-antiplasmin | P08697 |
| Alpha-2-HS-glycoprotein | P02765 |
| Alpha-2-macroglobulin | P01023 |
| Antithrombin-III | P01008 |
| Apolipoprotein A-I | P02647 |
| Apolipoprotein A-II | P02652 |
| Apolipoprotein A-IV | P06727 |
| Apolipoprotein B-100 | P04114 |
| Apolipoprotein C-I | P02654 |
| Apolipoprotein C-II | P02655 |
| Apolipoprotein C-III | P02656 |
| Apolipoprotein C-IV | P55056 |
| Apolipoprotein D | P05090 |
| Apolipoprotein E | P02649 |
| Apolipoprotein L1 | O14791 |
| Apolipoprotein M | O95445 |
| Apolipoprotein(a) | P08519 |
| Attractin | O75882 |
| Beta-2-glycoprotein 1 | P02749 |
| Beta-Ala-His dipeptidase | Q96KN2 |
| Biotinidase | P43251 |
| Cadherin-13 | P55290 |
| Carbonic anhydrase 1 | P00915 |
| Carboxypeptidase B2 | Q96IY4 |
| Cathelicidin antimicrobial peptide | P49913 |
| Cation-independent mannose-6-phosphate receptor | P11717 |
| CD5 antigen-like | O43866 |
| Ceruloplasmin | P00450 |
| Cholinesterase | P06276 |
| Clusterin | P10909 |
| Coagulation factor IX | P00740 |
| Coagulation factor V | P12259 |
| Coagulation factor VIII | P00451 |
| Coagulation factor X | P00742 |
| Coagulation factor XII | P00748 |
| Complement C1q subcomponent subunit B | P02746 |
| Complement C1q subcomponent subunit C | P02747 |
| Complement C1r subcomponent | P00736 |
| Complement C1r subcomponent-like protein | Q9NZP8 |
| Complement C1s subcomponent | P09871 |
| Complement C2 | P06681 |
| Complement C3 | P01024 |
| Complement C5 | P01031 |
| Complement component C7 | P10643 |
| Complement component C9 | P02748 |
| Complement factor B | P00751 |
| Complement factor H | P08603 |
| Complement factor I | P05156 |
| Corticosteroid-binding globulin | P08185 |
| Cystatin-C | P01034 |
| Endothelial protein C receptor | Q9UNN8 |
| Fetuin-B | Q9UGM5 |
| Fibrinogen alpha chain | P02671 |
| Fibrinogen beta chain | P02675 |
| Fibrinogen gamma chain | P02679 |
| Fibronectin | P02751 |
| Fibulin-1 | P23142 |
| Galectin-3-binding protein | Q08380 |
| Gelsolin | P06396 |
| Glutathione peroxidase 3 | P22352 |
| Haptoglobin | P00738 |
| Hemoglobin subunit alpha | P69905 |
| Hemopexin | P02790 |
| Heparin cofactor 2 | P05546 |
| Hepatocyte growth factor-like protein | P26927 |
| Hyaluronan-binding protein 2 | Q14520 |
| Ig mu chain C region | P01871 |
| Insulin-like growth factor I | P05019 |
| Inter-alpha-trypsin inhibitor heavy chain H2 | P19823 |
| Intercellular adhesion molecule 1 | P05362 |
| Interleukin-10 | P22301 |
| Kallistatin | P29622 |
| Keratin type I cytoskeletal 10 | P13645 |
| Keratin-type II cytoskeletal 2 epidermal | P35908 |
| Kininogen-1 | P01042 |
| Leucine-rich alpha-2-glycoprotein | P02750 |
| Lipopolysaccharide-binding protein | P18428 |
| L-selectin | P14151 |
| Lysozyme C | P61626 |
| Mannan-binding lectin serine protease 1 | P48740 |
| Mannan-binding lectin serine protease 2 | O00187 |
| Metalloproteinase inhibitor 2 | P16035 |
| Myeloblastin | P24158 |
| Peroxiredoxin-2 | P32119 |
| Phosphatidylinositol-glycan-specific phospholipase D | P80108 |
| Phospholipid transfer protein | P55058 |
| Pigment epithelium-derived factor | P36955 |
| Plasma protease C1 inhibitor | P05155 |
| Plasma serine protease inhibitor | P05154 |
| Plasminogen | P00747 |
| Plasminogen activator inhibitor 1 | P05121 |
| Pregnancy zone protein | P20742 |
| Protein AMBP | P02760 |
| Protein S100-A9 | P06702 |
| Protein Z-dependent protease inhibitor | Q9UK55 |
| Prothrombin | P00734 |
| Retinol-binding protein 4 | P02753 |
| Serotransferrin | P02787 |
| Serum albumin | P02768 |
| Serum paraoxonase/arylesterase 1 | P27169 |
| Serum paraoxonase/lactonase 3 | Q15166 |
| SPARC | P09486 |
| Tenascin | P24821 |
| Tenascin-X | P22105 |
| Thrombospondin-1 | P07996 |
| Thrombospondin-4 | P35443 |
| Thyroxine-binding globulin | P05543 |
| Tissue factor pathway inhibitor (isoform 1) | P10646 |
| Tissue-type plasminogen activator | P00750 |
| Transferrin receptor protein 1 | P02786 |
| Transthyretin | P02766 |
| Vascular cell adhesion protein 1 | P19320 |
| Vasorin | Q6EMK4 |
| Vitamin K-dependent protein S | P07225 |
| Vitamin K-dependent protein Z | P22891 |
| Vitronectin | P04004 |
| Zinc-alpha-2-glycoprotein | P25311 |
| Protein Name | UniProt Accession Number |
|---|---|
| 60 kDa heat shock protein mitochondrial | P10809 |
| 72 KDa type IV collagenase | P08253 |
| 78 kDa glucose-regulated protein | P11021 |
| A disintegrin and metalloproteinase with thrombospondin motifs 2 | O95450 |
| A disintegrin and metalloproteinase with thrombospondin motifs 20 | P59510 |
| A disintegrin and metalloproteinase with thrombospondin motifs 9 | Q9P2N4 |
| Actin, aortic smooth muscle | Actin, alpha cardiac muscle 1 | 8 other actin and actin-like proteins | P62736|P68032|8 others |
| Adhesion G protein-coupled receptor F5 | Q8IZF2 |
| Adipocyte plasma membrane-associated protein | Q9HDC9 |
| Adiponectin | Q15848 |
| ADM | P35318 |
| Afamin | P43652 |
| Alpha-1-acid glycoprotein 1 | P02763 |
| Alpha-1-antichymotrypsin | P01011 |
| Alpha-1-antitrypsin | P01009 |
| Alpha-1B-glycoprotein_VAR_018369 | P04217 |
| Alpha-2-antiplasmin | P08697 |
| Alpha-2-HS-glycoprotein | P02765 |
| Alpha-2-macroglobulin | P01023 |
| Angiogenin | P03950 |
| Angiopoietin-related protein 3 | Q9Y5C1 |
| Angiotensinogen | P01019 |
| Antithrombin-III | P01008 |
| Apolipoprotein A-I | P02647 |
| Apolipoprotein A-II | P02652 |
| Apolipoprotein A-IV | P06727 |
| Apolipoprotein B-100 | P04114 |
| Apolipoprotein C-I | P02654 |
| Apolipoprotein C-II | P02655 |
| Apolipoprotein C-III | P02656 |
| Apolipoprotein C-IV | P55056 |
| Apolipoprotein D | P05090 |
| Apolipoprotein E | P02649 |
| Apolipoprotein F | Q13790 |
| Apolipoprotein L1 | O14791 |
| Apolipoprotein M | O95445 |
| Apolipoprotein(a) | P08519 |
| Aromatase | P11511 |
| Atrial natriuretic peptide receptor 1 | P16066 |
| Attractin | O75882 |
| Autism susceptibility gene 2 protein | Q8WXX7 |
| B-cell scaffold protein with ankyrin repeats | Q8NDB2 |
| Beta-2-glycoprotein 1 | P02749 |
| Beta-2-microglobulin | P61769 |
| Beta-Ala-His dipeptidase | Q96KN2 |
| Beta-nerve growth factor | P01138 |
| Biotinidase | P43251 |
| C-reactive protein | P02741 |
| C4b-binding protein alpha chain | P04003 |
| Cadherin-13 | P55290 |
| Cadherin-5 | P33151 |
| Calcitonin | P01258 |
| Calcitonin gene-related peptide 1 | P06881 |
| Calponin-1 | P51911 |
| Carbonic anhydrase 1 | P00915 |
| Carboxypeptidase B2 | Q96IY4 |
| Carboxypeptidase N catalytic chain | P15169 |
| Carboxypeptidase N subunit 2 | P22792 |
| Cartilage acidic protein 1 | Q9NQ79 |
| Cathelicidin antimicrobial peptide | P49913 |
| Cation-independent mannose-6-phosphate receptor | P11717 |
| CD40 ligand | P29965 |
| CD44 antigen | P16070 |
| CD5 antigen-like | O43866 |
| cDNA FLJ53327, highly similar to Gelsolin | B7Z2X4 |
| Ceruloplasmin | P00450 |
| Cholesteryl ester transfer protein | P11597 |
| Cholinesterase | P06276 |
| Chromogranin-A | P10645 |
| Claudin-5 | O00501 |
| Clusterin | P10909 |
| Coagulation factor IX | P00740 |
| Coagulation factor V | P12259 |
| Coagulation factor VII | P08709 |
| Coagulation factor VIII | P00451 |
| Coagulation factor X | P00742 |
| Coagulation factor XI | P03951 |
| Coagulation factor XII | P00748 |
| Coagulation factor XIII A chain | P00488 |
| Coagulation factor XIII B chain | P05160 |
| Collagen alpha-1(I) chain | P02452 |
| Collagen alpha-1(III) chain | P02461 |
| Collagen alpha-1(XVIII) chain | P39060 |
| Collagen alpha-2(I) chain | P08123 |
| Complement C1q subcomponent subunit A | P02745 |
| Complement C1q subcomponent subunit B | P02746 |
| Complement C1q subcomponent subunit C | P02747 |
| Complement C1r subcomponent | P00736 |
| Complement C1r subcomponent-like protein | Q9NZP8 |
| Complement C1s subcomponent | P09871 |
| Complement C2 | P06681 |
| Complement C3 | P01024 |
| Complement C5 | P01031 |
| Complement component C6 | P13671 |
| Complement component C7 | P10643 |
| Complement component C8 alpha chain | P07357 |
| Complement component C8 beta chain | P07358 |
| Complement component C9 | P02748 |
| Complement factor B | P00751 |
| Complement factor D | P00746 |
| Complement factor H | P08603 |
| Complement factor I | P05156 |
| Complement C4 | P0C0L4|P0C0L5 |
| Corticosteroid-binding globulin | P08185 |
| Creatine kinase B-type | P12277 |
| Creatine kinase M-type | P06732 |
| Cystatin-C | P01034 |
| Desmoplakin | P15924 |
| Di-N-acetylchitobiase | Q01459 |
| Dickkopf-related protein 1 and 2 | O94907|Q9UBU2 |
| E-selectin | P16581 |
| Elastin | P15502 |
| Endothelial lipase | Q9Y5X9 |
| Endothelial protein C receptor | Q9UNN8 |
| Epidermal growth factor receptor | P00533 |
| Extracellular matrix protein 1 | Q16610 |
| Fatty acid-binding protein heart | P05413 |
| Ferritin heavy chain | P02794 |
| Ferritin light chain | P02792 |
| Fetuin-B | Q9UGM5 |
| Fibrinogen alpha chain | P02671 |
| Fibrinogen beta chain | P02675 |
| Fibrinogen gamma chain | P02679 |
| Fibronectin | P02751 |
| Fibulin-1 | P23142 |
| Ficolin-2 | Q15485 |
| Ficolin-3 | O75636 |
| Follistatin-related protein 1 | Q12841 |
| Fructose-bisphosphate aldolase B | P05062 |
| Galectin-3 | P17931 |
| Galectin-3-binding protein | Q08380 |
| Gamma-enolase | P09104 |
| Gelsolin | P06396 |
| Glial fibrillary acidic protein | P14136 |
| Glutamate receptor ionotropic NMDA 2A | Q12879 |
| Glutamate receptor ionotropic NMDA 2B | Q13224 |
| Glutathione peroxidase 3 | P22352 |
| Glutathione S-transferase P | P09211 |
| Haptoglobin | P00738 |
| Heat shock protein beta-1 | P04792 |
| Hemoglobin subunit alpha | P69905 |
| Hemopexin | P02790 |
| Heparin cofactor 2 | P05546 |
| Hepatocyte growth factor-like protein | P26927 |
| Histidine-rich glycoprotein | P04196 |
| Hornerin | Q86YZ3 |
| Hyaluronan-binding protein 2 | Q14520 |
| Ig gamma-1 chain C region | P01857 |
| Ig mu chain C region | P01871 |
| Ig mu heavy chain disease protein and Ig mu chain C | P04220|P01871 |
| IgGFc-binding protein | Q9Y6R7 |
| Immunoglobulin kappa variable 4-1 | P06312 |
| Insulin-like growth factor I | P05019 |
| Insulin-like growth factor-binding protein 1 | P08833 |
| Insulin-like growth factor-binding protein 2 | P18065 |
| Insulin-like growth factor-binding protein 3 | P17936 |
| Insulin-like growth factor-binding protein complex acid labile subunit | P35858 |
| Inter-alpha-trypsin inhibitor heavy chain H1 | P19827 |
| Inter-alpha-trypsin inhibitor heavy chain H2 | P19823 |
| Inter-alpha-trypsin inhibitor heavy chain H4 | Q14624 |
| Intercellular adhesion molecule 1 | P05362 |
| Interleukin-10 | P22301 |
| Interleukin-6 | P05231 |
| Interstitial collagenase | P03956 |
| Kallistatin | P29622 |
| Keratin type I cytoskeletal 10 | P13645 |
| Keratin type I cytoskeletal 9 | P35527 |
| Keratin-type II cytoskeletal 2 epidermal | P35908 |
| Kininogen-1 | P01042 |
| L-selectin | P14151 |
| Lactotransferrin | P02788 |
| Leucine-rich alpha-2-glycoprotein | P02750 |
| Lipopolysaccharide-binding protein | P18428 |
| Lumican | P51884 |
| Lysozyme C | P61626 |
| Mannan-binding lectin serine protease 1 | P48740 |
| Mannan-binding lectin serine protease 2A | O00187 |
| Mannose-binding protein C | P11226 |
| Matrix Gla protein | P08493 |
| Matrix metalloproteinase-9 | P14780 |
| Melanotransferrin | P08582 |
| Metalloproteinase inhibitor 1 | P01033 |
| Metalloproteinase inhibitor 2 | P16035 |
| Metalloproteinase inhibitor 4 | Q99727 |
| Microtubule-associated protein tau | P10636 |
| Mucin-16 | Q8WXI7 |
| Myelin basic protein | P02686 |
| Myeloblastin | P24158 |
| Myeloperoxidase | P05164 |
| N-acetylmuramoyl-L-alanine amidase | Q96PD5 |
| N(G),N(G)-dimethylarginine dimethylaminohydrolase 1 | O94760 |
| Natriuretic peptides B | P16860 |
| Neuropilin-2 | O60462 |
| Neutrophil gelatinase-associated lipocalin | P80188 |
| Nucleoside diphosphate kinase A | P15531 |
| Occludin | Q16625 |
| Osteopontin | P10451 |
| Oxidized low-density lipoprotein receptor 1 | P78380 |
| P-selectin | P16109 |
| Pappalysin-1 | Q13219 |
| Peroxiredoxin-1 | Q06830 |
| Peroxiredoxin-2 | P32119 |
| Phosphatidylcholine-sterol acyltransferase | P04180 |
| Phosphatidylinositol-glycan-specific phospholipase D | P80108 |
| Phospholipid transfer protein | P55058 |
| Pigment epithelium-derived factor | P36955 |
| Plasma protease C1 inhibitor | P05155 |
| Plasma serine protease inhibitor | P05154 |
| Plasminogen | P00747 |
| Plasminogen activator inhibitor 1 | P05121 |
| Plastin-2 | P13796 |
| Platelet endothelial cell adhesion molecule | P16284 |
| Platelet glycoprotein VI | Q9HCN6 |
| Platelet-activating factor acetylhydrolase | Q13093 |
| Pregnancy zone protein | P20742 |
| Proenkephalin-A | P01210 |
| Prolactin | P01236 |
| Protein AMBP | P02760 |
| Protein S100-A12 | P80511 |
| Protein S100-A9 | P06702 |
| Protein S100-B | P04271 |
| Protein Z-dependent protease inhibitor | Q9UK55 |
| Protein_deglycase_DJ-1 | Q99497 |
| Proteoglycan 4 | Q92954 |
| Prothrombin | P00734 |
| Ras GTPase-activating protein nGAP | Q9UJF2 |
| Resistin | Q9HD89 |
| Retinol-binding protein 4 | P02753 |
| Serotransferrin | P02787 |
| Serum albumin | P02768 |
| Serum amyloid A-1 and A-2 proteins | P0DJI8|P0DJI9 |
| Serum amyloid A-4 protein | P35542 |
| Serum amyloid P-component | P02743 |
| Serum paraoxonase/arylesterase 1 | P27169 |
| Serum paraoxonase/lactonase 3 | Q15166 |
| Sex hormone-binding globulin | P04278 |
| SPARC | P09486 |
| Spermine oxidase | Q9NWM0 |
| Sterile alpha motif domain-containing protein 9-like | Q8IVG5 |
| Stromelysin-1 | P08254 |
| Target of Nesh-SH3 | Q7Z7G0 |
| TBC1 domain family member 10A | Q9BXI6 |
| Tenascin | P24821 |
| Tenascin-X | P22105|Q16473 |
| Tetranectin | P05452 |
| Thrombomodulin | P07204 |
| Thrombospondin-1 | P07996 |
| Thrombospondin-4 | P35443 |
| Thyroglobulin | P01266 |
| Thyroxine-binding globulin | P05543 |
| Tissue_factor_pathway_inhibitor | P10646 |
| Tissue-type plasminogen activator | P00750 |
| Transcription factor SOX-11 | P35716 |
| Transferrin receptor protein 1 | P02786 |
| Transthyretin | P02766 |
| Tumor necrosis factor receptor superfamily member 1A | P19438 |
| Tumor necrosis factor receptor superfamily member 1B | P20333 |
| Vascular cell adhesion protein 1 | P19320 |
| Vascular endothelial growth factor B | P49765 |
| Vascular endothelial growth factor D | O43915 |
| Vascular non-inflammatory molecule 3 | Q9NY84 |
| Vasorin | Q6EMK4 |
| Vitamin D-binding protein | P02774 |
| Vitamin K-dependent protein C | P04070 |
| Vitamin K-dependent protein S | P07225 |
| Vitamin K-dependent protein Z | P22891 |
| Vitronectin | P04004 |
| von Willebrand factor | P04275 |
| Xaa-Pro dipeptidase | P12955 |
| Zinc-alpha-2-glycoprotein | P25311 |