The Critical Challenge of Model Validation in Alzheimer’s Drug Development
The pharmaceutical industry faces a persistent challenge in neurodegenerative disease research: ensuring that preclinical models accurately recapitulate the complex biochemical landscape of human disease. For Alzheimer’s disease (AD), where clinical trial failure rates exceed 99%, the stakes of model selection have never been higher. While traditional approaches to validating animal models have relied on histopathological markers and behavioral assessments, these methods often fail to capture the intricate metabolic dysregulation that underlies disease progression. The question that confronts every research team initiating an Alzheimer’s program is whether their chosen model system truly reflects the biochemical perturbations occurring in patients, or whether they are studying an artificial phenotype that will not translate to human therapeutic benefit.
Recent advances in metabolic signatures of Alzheimer’s Disease have provided a powerful new lens through which researchers can evaluate the biological fidelity of preclinical models. Next Generation Metabolomics® profiling offers an unbiased, comprehensive view of the biochemical state of a biological system, capturing both upstream driver pathways and downstream consequences of disease processes. When applied to model validation, this technology enables researchers to move beyond single-marker assessments and instead compare the entire metabolic landscape of their model system against known human disease signatures.
Why is ApoE Important in Alzheimer’s Disease?
Apolipoprotein E stands as the most significant genetic risk factor for late-onset Alzheimer’s disease, with the Apo ε4 allele conferring substantially elevated risk compared to the more common ε3 variant (Figure 1).
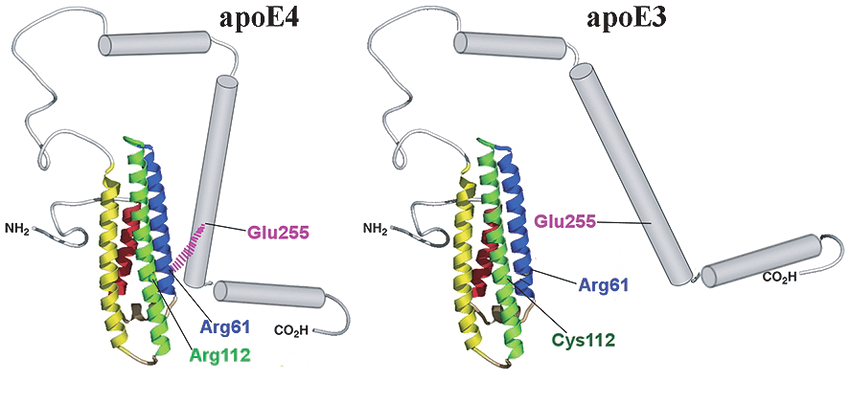
Figure 1: Structural differences of Apo ε4 and ApoE3 (doi:10.3233/JAD-2010-100009)
Beyond its role in lipid transport, ApoE profoundly influences multiple processes implicated in AD pathogenesis, including amyloid-beta clearance, synaptic integrity, neuroinflammation, and blood-brain barrier function. The ApoE knockout model has emerged as a valuable tool for dissecting these mechanisms, exhibiting synaptic damage, cholinergic dysfunction, and cognitive impairment consistent with early Alzheimer’s pathology.
However, the metabolic consequences of ApoE deficiency and their relevance to human disease have remained incompletely characterized. This gap represents a critical limitation for drug development programs utilizing these models, as metabolic dysregulation may represent both a key driver of pathology and a sensitive readout of therapeutic efficacy. Without comprehensive metabolic validation, researchers risk pursuing compounds that appear efficacious in their model system but fail to engage the metabolic pathways disrupted in human patients.
Metabolomic Validation of ApoE Knockout Rat Models
A systematic metabolomic analysis of ApoE knockout rats compared to wild-type controls has revealed striking parallels between the metabolic phenotype of these animals and documented biochemical abnormalities in Alzheimer’s disease. Using liquid chromatography-mass spectrometry (LC/MS) to profile over 3,000 metabolites across liver and serum samples, researchers identified 26 serum metabolites and 46 liver metabolites that showed statistically significant correlation with ApoE status. Perhaps more importantly, pathway enrichment analysis (Figure 2) revealed that these differential metabolites cluster within specific biochemical pathways known to be dysregulated in human AD, providing systems-level validation of the model’s disease relevance.

Figure 2: Pathway enrichment analysis of liver (left) and serum (right) metabolomics data. The dot plots show enriched pathways and their significance (x-axis), with the p = 0.05 threshold marked by a gray dashed line. Dot size indicates the number of significantly different metabolites, and color represents the enrichment ratio.
The global metabolic shift (Figure 3) observed in both peripheral tissues of ApoE knockout animals suggests that the consequences of ApoE deficiency extend far beyond the central nervous system. This finding aligns with emerging evidence that Alzheimer’s disease involves systemic metabolic dysfunction rather than being purely a brain disorder. For drug development programs, this systemic metabolic phenotype offers the potential to use peripheral biomarker discovery as a window into central therapeutic effects, potentially accelerating the development timeline by enabling pharmacodynamic monitoring without requiring cerebrospinal fluid sampling or PET imaging.
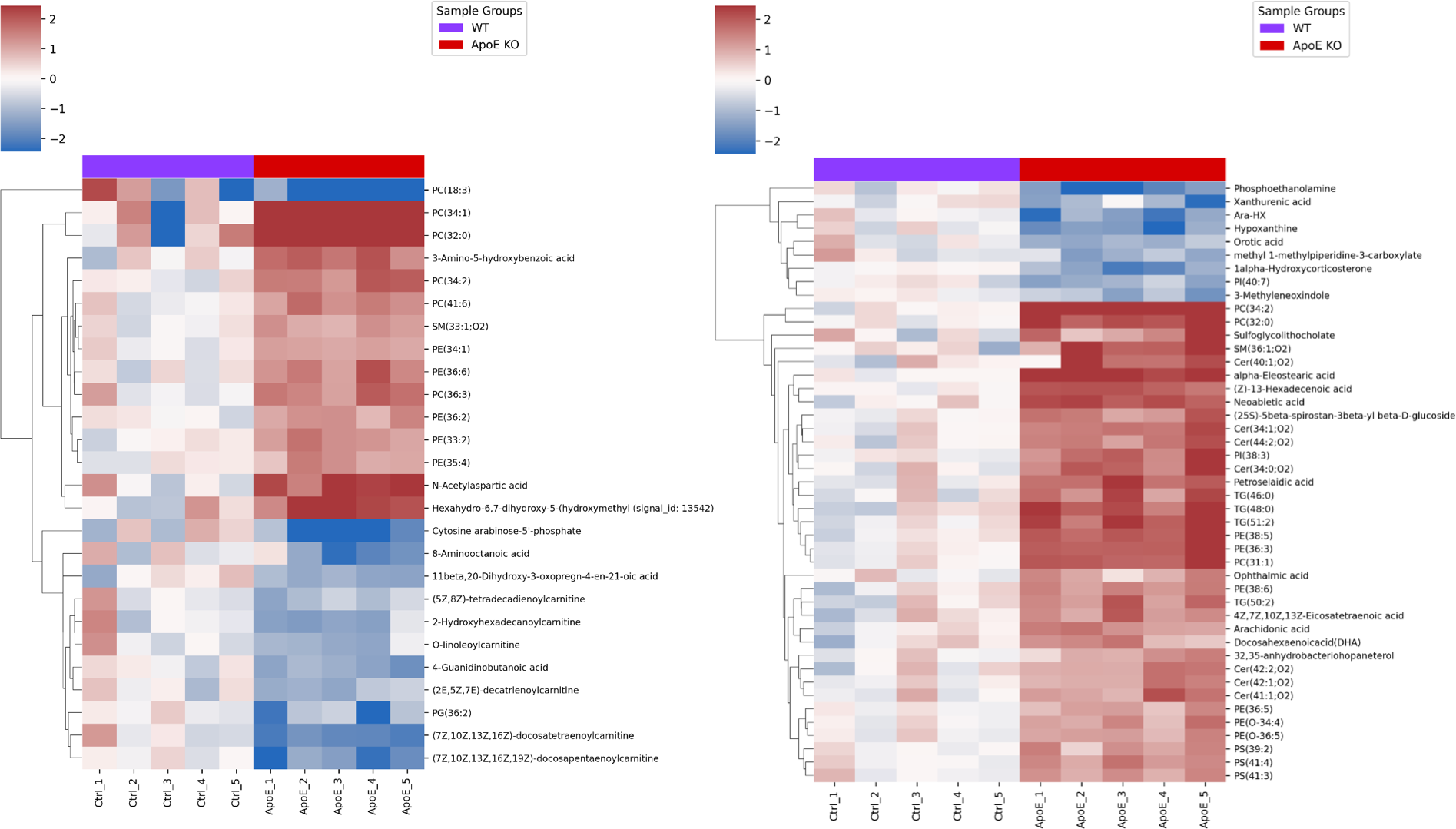
Figure 3: Heatmaps of liver and serum metabolites show a statistically significant correlation with ApoE status. Heatmaps show the log2(fc) relative to the mean level across the samples. Columns are colored according to ApoE status.
Mitochondrial Dysfunction and Fatty Acylcarnitine Depletion
One of the most striking findings from the metabolomic analysis was the significant depletion of fatty acylcarnitines in the liver of ApoE knockout animals. These molecules serve as essential intermediates in the carnitine shuttle system, which transports long-chain fatty acids across the mitochondrial membrane for beta-oxidation. The reduction in fatty acylcarnitine pool sizes suggests impaired mitochondrial fuel delivery, potentially compromising the cellular capacity for energy production through fatty acid oxidation.
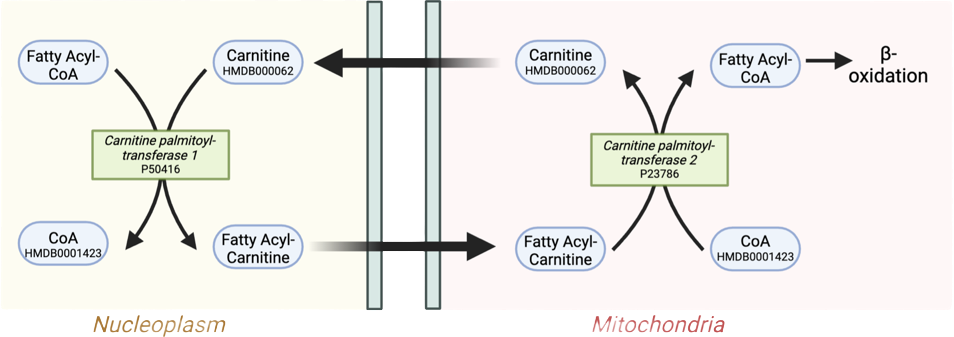
Figure 4: Diagram of carnitine shuttle. Fatty acyls are transported into the mitochondria after conversion to fatty acylcarnitines, allowing them to enter the β-oxidation pathway for energy production.
This finding carries particular significance because mitochondrial dysfunction represents a well-established feature of Alzheimer’s disease, with multiple studies documenting decreased mitochondrial respiration, altered mitochondrial morphology, and disrupted energy metabolism in affected brain regions. The observation of disturbed carnitine metabolism and decreased acylcarnitine levels in AD patients mirrors the pattern observed in ApoE knockout rats, providing metabolic validation that these animals recapitulate a key energetic deficit of the human disease. For researchers developing therapies targeting mitochondrial function or metabolic support pathways, this correspondence suggests that the ApoE knockout model may provide a relevant system for evaluating compound efficacy.
The mechanistic implications extend beyond simple energy deficiency. Carnitine-deficient states lead to the accumulation of fatty acyl-CoA species in the cytoplasm, potentially triggering lipotoxic stress responses and inflammatory signaling cascades. These secondary consequences of impaired fatty acid oxidation may contribute to the neurodegenerative cascade, suggesting that interventions supporting mitochondrial fuel utilization could yield therapeutic benefit through multiple mechanisms.
Sphingolipid Dysregulation and Ceramide Elevation
Perhaps the most disease-relevant finding from the metabolomic analysis was the pronounced dysregulation of sphingolipid metabolism in ApoE knockout animals (Figure 5). Pathway enrichment analysis revealed significant alterations in sphingolipid-related pathways in both serum and liver, with eight specific ceramide species showing elevated levels in knockout animals compared to wild-type controls. This metabolic signature closely parallels extensive documentation of altered sphingolipid metabolism and increased ceramide levels in Alzheimer’s disease patients, where elevated brain and serum ceramides have been associated with disease risk, progression, and severity.
The biological significance of ceramide elevation in neurodegenerative disease models extends well beyond simple biomarker correlation. Ceramides play active roles in multiple pathological processes central to Alzheimer’s pathogenesis, including the promotion of amyloid-beta peptide production through effects on secretase activity, the induction of neuronal apoptosis through activation of stress-activated protein kinases, and potential involvement in tau hyperphosphorylation. The observation that ApoE knockout rats spontaneously develop this sphingolipid dysregulation – without genetic manipulation of ceramide synthesis pathways or dietary interventions – suggests that the model naturally engages in disease-relevant lipid metabolism pathways.
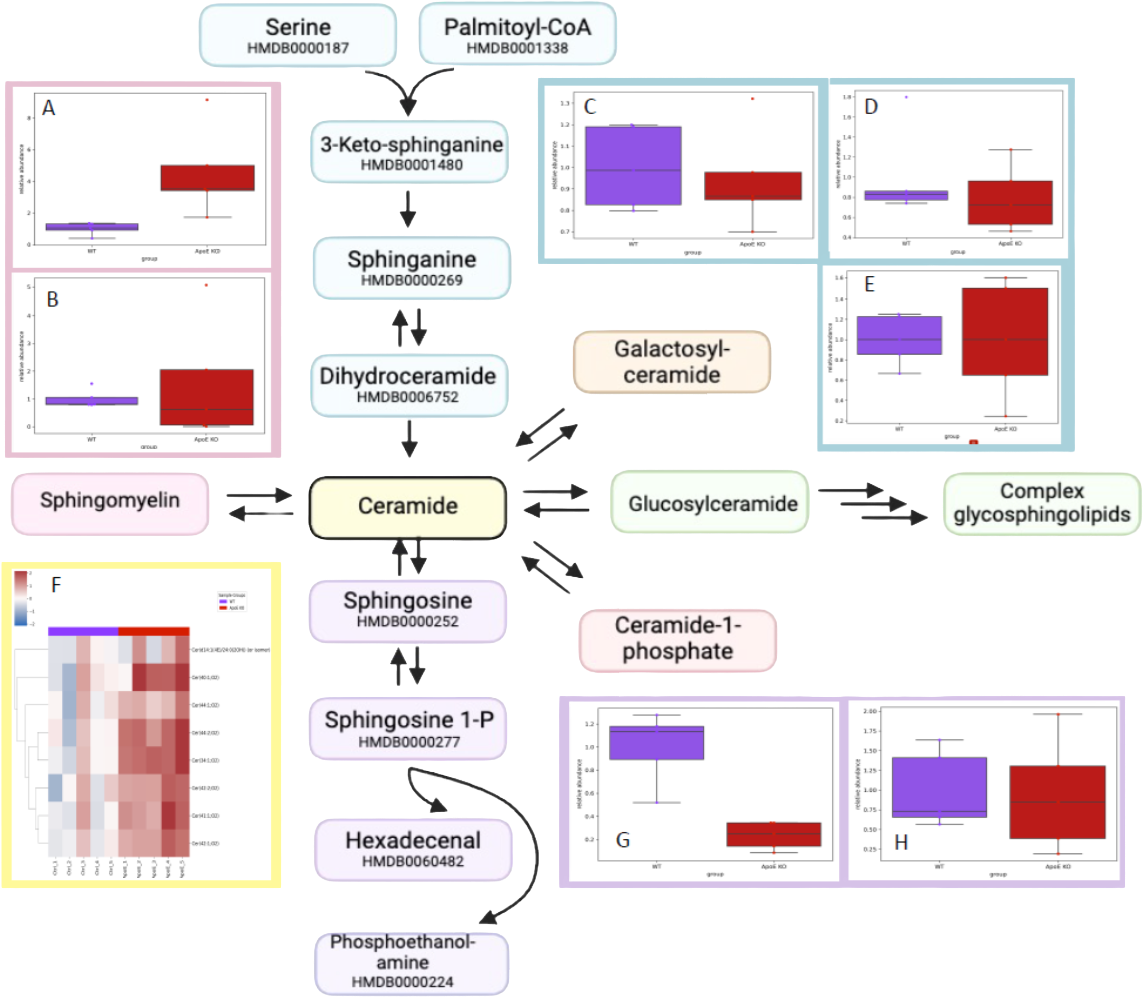
Figure 5: Sphingolipid metabolism pathways are shown. (A–B) Sphingomyelinase (pink): SM(36:1;O2) was enriched in ApoE KO, SM(d17:1/26:1)* was not. C–E) De novo (blue): no significant differences. F) Ceramide heatmap: 8 ceramides enriched in ApoE KO (log2 fold change). G) Catabolic (purple): phosphoethanolamine was enriched, sphingosine 1-P* was not (*or isomer).
For preclinical Alzheimer’s research programs, this sphingolipid phenotype provides multiple opportunities for therapeutic intervention and biomarker development. Compounds targeting ceramide synthesis, sphingomyelin hydrolysis, or ceramide clearance pathways could be evaluated for their ability to normalize this metabolic signature while simultaneously assessing effects on cognitive and neuropathological outcomes. The peripheral accessibility of ceramide measurements in serum offers the potential for pharmacodynamic monitoring throughout preclinical studies, providing early evidence of target engagement before investing in more resource-intensive histological or behavioral endpoints.
Translational Implications for Alzheimer’s Drug Development
The metabolomic validation of ApoE knockout rats carries important implications for multiple stages of the drug development pipeline. At the earliest stages of program initiation, these data provide evidence-based support for model selection decisions, particularly for programs targeting metabolic dysfunction, mitochondrial support, or lipid metabolism in Alzheimer’s. The demonstration that ApoE knockout animals recapitulate multiple metabolic abnormalities documented in human AD patients reduces the risk that efficacy signals observed in these models represent artifacts of the experimental system rather than genuine therapeutic effects likely to translate to human benefit.
For ongoing research programs, comprehensive metabolomic profiling of model systems enables more sophisticated experimental design and data interpretation. Rather than relying solely on traditional efficacy endpoints like amyloid burden or cognitive testing, researchers can incorporate metabolic biomarkers as intermediate pharmacodynamic readouts, potentially identifying promising compounds earlier in the screening process or gaining mechanistic insights into why certain interventions succeed or fail. The identification of dysregulated pathways in the model system also suggests rational combination therapy approaches, where metabolic support could be combined with other therapeutic modalities to achieve synergistic benefit.
The system’s biology perspective enabled by metabolomic profiling also helps address a persistent challenge in translational neuroscience: understanding which aspects of a complex animal model are most relevant to human disease. Not all features of any given model system will perfectly mirror human pathology, and distinguishing disease-relevant phenotypes from model-specific artifacts require careful biochemical characterization. The convergence between ApoE knockout rat metabolic signatures and documented human Alzheimer’s disease metabolomic abnormalities provides confidence that observed effects in this model are likely to reflect processes occurring in patients rather than rodent-specific biology.
Conclusion: Metabolomics as a Model Validation Tool
The comprehensive metabolomic characterization of ApoE knockout rats demonstrates the power of unbiased biochemical profiling to validate preclinical disease models at a systems level. By revealing that these animals recapitulate key metabolic abnormalities documented in human Alzheimer’s disease, including fatty acylcarnitine depletion indicative of mitochondrial dysfunction and ceramide elevation reflecting sphingolipid dysregulation – this analysis provides strong support for the continued use of ApoE-deficient models in neurodegenerative research. More broadly, this work illustrates how metabolomic approaches can inform model selection, enhance experimental design, and ultimately improve the probability of translational success for Alzheimer’s disease drug development programs.
For research teams seeking to validate their own model systems or gain deeper mechanistic insights into disease biology, Panome Bio’s Next Generation Metabolomics services offer the comprehensive, unbiased profiling platform necessary to characterize complex disease states at the molecular level. As the field continues to grapple with the challenge of improving clinical trial success rates, the integration of advanced metabolomic technologies into preclinical research represents an important step toward developing therapeutics that address the true biochemical dysfunction underlying human disease.